The University of Texas Southwestern Medical Center at Dallas
ADULT-ONSET STILLS DISEASE
A Circadian Cytokine Syndrome?
John J. Cush, MD
Internal Medecine Grand Rounds
5 January 1994
Introduction
Adult-onset Still's disease (AOSD) is a recently recognized systemic inflammatory disorder of unknown etiology and pathogenesis. Although this diagnosis was popularized by a 1971 report by Sir Eric Bywaters, similar cases have been sporadically reported thorough out this century. AOSD is likely to be the adult continuum of the better known systemic-onset juvenile arthritis 0 7,21,94). Characteristic manifestations include quotidian fever, evanescent rash, sore throat, arthritis, polyserositis, leukocytosis and seronegativity. Its clinical course is marked by systemic exacerbations and/or chronic arthritis.
There is no diagnostic test or pathognomonic histopathology and thus, the diagnosis of AOSD is often problematic. Early reports diagnosed AOSD in patients with long standing history of fever, arthritis and multiple systemic complaints. A large proportion of documented patients have suffered from extended delays in diagnosis due to protracted, expansive and often costly efforts to exclude occult infections or neoplasms. By contrast, AOSD is now considered after several week so in the evaluation of undiagnosed fever and rheumatic complaints. AOSD remains a painstakingly difficult clinical diagnosis, largely because of its rarity, protean manifestations and a lack of pathognomic features or diagnostic tests. This review will acquaint the clinician with this syndrome and provide an approach to these problems based on personal experience and information from the worlds literature.
Juvenile Arthritis (JA)
Juvenile arthritis WA) is also known as Juvenile Rheumatoid Arthritis WRA), Juvenile Chronic Polyarthritis or Still's Disease. Although the latter term is often broadly used in Europe to include all forms of JA, the term "Still's Disease" in refers to the systemic variant of JA that was described by George F. Still in 1897.
The growth of pediatric rheumatology as a subspecialty has enhanced our understanding of JA. Juvenile arthritis comprises several entities unified by the presence of a chronic inflammatory arthropathy with its onset before the age of 16 years (Ansell 1991, Fink 1983). These subsets are detailed below and are distinguished by the degree of articular involvement, onset age and other distinctive features (Table 1).
Pauciarticular-onset JA refers to involvement of 4 or fewer joints in the first 6 weeks, while polyarticular disease requires that 5 or more joints be involved at the onset and often continues throughout the disease. The pauciarticular group constitutes the vast majority of juvenile patients with arthritis and is subdivided into 2 types. Type I Pauciarticular JA comprises the largest percentage of JA patient and has a younger onset and a relatively good articular prognosis. A significant number of these children are at risk to develop iridocyclitis (uveitis) that may correlate with the presence of antinuclear antibodies (usually in low titer), but not autoantibodies to native DNA. A minority of pauciarticular-onset children will manifest Type II Pauciarticular JA. These children tend to be older-onset (> 9 years) males, who are often HLA-B27 positive and present with large joint monarticular disease, hip or spinal involvement. This subset has also been labeled juvenile spondylitis and includes children with early ankylosing spondylitis, Reiter's syndrome, psoriatic arthritis (PsA), or inflammatory bowel disease (IBD) associated arthropathy. In the juvenile spondylitis population, arthritis usually precedes the onset of extra-articular disease (ie, enthesitis, uveitis, nail pitting, psoriatic plaques, diarrhea, etc.). Such patients may have a family history of AS, RS, IBD or psoriasis.
Polyarticular JA is seen in a minority of patients and often presents as a symmetric inflammatory polyarticular process with a propensity for chronicity and significant disability. Up to 10% of JA patients will be seropositive for IgM rheumatoid factor (RF) and are considered to be the juvenile continuum of adult rheumatoid arthritis and behave as such. The larger number of seronegative polyarticular JA patients are also at risk for aggressive joint disease, and should be monitored closely and treated aggressively.
| Variant (onset type) | %JRA | Age | Sex | Clinical features | ANA | RF | HLA | Prognosis |
| Pauciarticular - Type I | 50% | 1-8 | F | Few joints | 60% | 0 | DRS DR6 -DR8 | Joints OK -Uvéitis 30-50% |
| Pauciarticular - Type II | 10% | 9-16 | M | Few Joints uveitis, Sxs of Reiter's, Psoriatic or IBD arthritis | 0 | 0 | B27 | ? Risk of spondylitis |
| Polyarticular RF + | 5% | 10-16 | F | RA nodules weight loss | 70% | 100 | DR4 | Erosive arthritis >50% |
| Polyarticular RF- | 15% | 3-9 | F | Symmetric polyarthritis | 0 | 0 | Erosive arthritis 15 % | |
| Systemic Onset | 20% | 5-16 | M=F | Spiking fever, rash, serositis organomely anemia, WBC & ESR | 0 | 0 | Bw35 DR4 | Erosive arthritis 20% |
Table 1. JUVENILE ARTHRITIS - Distinguishing features
Systemic-onset JA is seen in roughly 20% of all JRA patients (Hafner 1986, Prieur 1984, Svantesson 1983). While this variant has been reported at all ages (up to age 16 years), the onset is most common between 5 and 15 years of age. Cardinal manifestations include high spiking, daily (quotidian) fevers, evanescent rash, arthritis, splenomegaly, lymphadenopathy, serositis, weight loss, neutrophilic leukocytosis, increased acute phase proteins and seronegative tests for ANA and RF. Up to 25% of these patients will develop a chronic symmetric polyarthritis that is indistinguishable from rheumatoid arthritis.
Similarity between systemic-onset JA and AOSD? Although frequently inferred, it is unknown if systemic-onset JA and AOSD are the same disease and share similar manifestations and clinical course. Neither systemic-onset JA nor AOSD have been ascribed an acceptable or unifying etiology to date. Nonetheless, despite the defined differences in onset age, both populations share a remarkable number of clinical features (Table 2). Few reports have attempted to compare these populations.
| FEATURE | SYSTEMIC-ONSET JA* | AOSD |
| Sex | M = F | M |
| F | ||
| Quotidian Fever | 99% | |
| 94% | ||
| Evanescent Rash | 90% | 87% |
| Arthritis | 95% | 93% |
| Prodromal Sore Throat | 15% | 70% |
| RES Involvement | 40-70% | |
| 50-70% | ||
| Serositis | 20-50% | 20-40% |
| Serologic Tests | ANA(-), RF(-) | ANA(-),RF(-) |
| Carpal Ankylosis | 28-50% | |
| 45-55% | ||
| Destructive Arthritis | 30 (20-60)% | 20-25% |
| HLA Associations | Bw35, DR2, DR4, DR5, Dw7 | Bw35, DR4, Dw7 |
* based on reports of Fernandez-Vina, Lang, Cabane, Calabro, Delpaine, Tanaka, Talesnik
Table 2. Comparison of Systemic-Onset Juvenile Arthritis and Adult-Onset Still's disease
In Japan, Tanaka and colleagues have shown comparable clinical manifestations, treatment responses and prognosis for 26 systemic-onset JA and 19 AOSD patients (Tanaka 1991).
The only disparate feature was the prodromal sore throat that was present in 68% of AOSD, but only 19% of systemic-onset JA patients. Cabane et al have shown that both juvenile and adult populations share a good prognosis with regard to systemic/extraarticular disease, but are at significant risk for chronic articular sequelae (Cabane 1990). Ultimately, 4/10 JA patients and 3/8 AOSD patients required total hip prosthesis because of advanced hip disease. The claim of problematic articular disease and sequelae has often been made for both juvenile (Cabane 1988, Mozziconacci 1983, Talesnik 1992) and adult populations (Cush 1985, Wouters 1986). Talesnik has also shown similar clinical courses when comparing 14 SOJA and 7 AOSD patients (Talesnik 1992). Nearly 50% of AOSD and systemic-onset JA patients both manifest chronic articular disease and that significant articular sequelae were only observed in these patients. Lastly, multiple investigators have demonstrated similar HLA associations (ie, HLA-Bw35 and DR4) for both populations (Miller, Wouters, Terkeltaub). Therefore, it appears that Still's disease is likely to be the same disorder in both children and adults. This notion is likely to prove valuable when considering data regarding etiology, pathogenesis, or therapeutic measures in Still's disease.
History
Juvenile arthritis was first described by George Frederick Still (1868-1941) who presented his thesis in an 1897 address entitled, "A special form of joint disease met with in children". This work was based on Still's clinical experience as a medical registrar and pathologist at The Hospital for Sick Children on Great Ormond Street in London. Still was the first to comprehensively describe the occurrence of a chronic arthritis in 22 children, 19 of whom he cared for. In fact, Still described three variants of arthritis found in children: rheumatoid arthritis, Jaccouds' arthropathy ("chronic fibrous rheumatism"), and the systemic-onset variant that still bears his name (Bywaters 1967). He described a chronic disorder of the joints that differed from adult rheumatoid arthritis and stressed the onset before the second
dentition, near equal female:male ratio, fever patterns,
lymphadenopathy, splenomegaly, pericardial and pleural inflammation, anemia, lack of
deformity and retardation of growth (Baum 1978, Birch 1973, Ansell 1991. Such a chronic
arthropathy was also described in adults by Bannatyne and Chauffard (1896) who each
reported patients with acute onset rheumatoid arthritis associated with fever,
lymphadenopathy and/or splenomegaly.
1896 - Bannatyne and Chauffard
1897 GF Still describe 22 JA pts
1933 Boldero: 1st:described rheumatoid rash ".
1933 MoItke : " Stills disease in adults ".
1943 - Wissler & Fanconi: " subsepsis hyperallergica "
1971 - Bywaters: 14 females with AOSD.
1973 Bujak et al: 10 males with AOSD.
1976 - Medsger et al : carpal ankylosis in AOSD
Table 3. History of Still's Disease
The distinctive evanescent rash, termed a
"rheumatoid rash" or "Still's rash" was not described by Still, but
instead by Boldero who, in 1933, described the characteristic evanescent erythematous,
rash involving the extensor surfaces (Boldero 1933). Commentary offered by Professor F.
Langmead suggested that pyrexial attacks were often associated with this rash. The
characteristics of this distinctive exanthem were further described by Isdale and
Bywaters, who demonstrated a strong association between the "rheumatoid rash"
and other characteristic features of Still's disease (e.g. intermittent fever,
lymphadenopathy, splenomegaly, leukocytosis, and an elevated erythrocyte sedimentation
rate) (Isdale 1957). They also described the occurrence of this rash in 7 of 500 adults
with rheumatoid arthritis. These 7 patients later comprised the basis for Bywaters 1971
report of Still's disease in the adult (Bywaters 1971).
Sporadic descriptions of patients with manifestations
similar to that described by Still are occasionally found and even predate Still's
original description. Nonetheless, in 1933 Otto Moltke published a report entitled
"Still's disease in adults". MoItke first questioned the arbitrary age limits
ascribed to JA and Still's disease and suggested that this affliction may occur up to 35
years of age (Moltke 1933). He described 4 young males, ages 1528, with an acute
polyarthritis, fever, profuse perspiration, lymphadenopathy, sore throat, muscular
atrophy, anemia and elevated erythrocyte sedimentation rates. Soon thereafter the 1940's
French literature popularized the "WisslerFanconi syndrome" and
"subsepsis hyperallergica" as terms to describe an analogous clinical picture of
AOSD in young adults (95). Numerous descriptions have since appeared in the English
literature describing similar clinical pictures without the label of AOSD. It wasn't until
the early 1970's that Bywaters (1971) and Bujak et al 1973) revealed two large,
independent clinical series establishing this juvenile entity as an adult affliction.
Finally, in 1976 Medsger and Christy described the frequency of carpal ankylosis in AOSD.
This distinctive pattern of arthritis along with the characteristic quotidian fevers and
evanescent rashes constitutes the triad of features upon which the diagnosis of AOSD
frequently rests.
Epidemiology
Reports of AOSD have appeared from
many countries, involving many races and therefore seems unlikely that a significant
racial geographic distribution. While all races have been affected, a majority of patients
thus far reported have been Caucasian, although a substantial number of Blacks, Orientals
and Latinos have been described. Although the number of published reports has increased
and enhanced the awareness of AOSD (Figure 1), it is a rarely encountered disorder. This
review details over 400 patients reported in the English and foreign literature since
1971, with nearly half of these being individual case reports. The rarity of this syndrome
is also suggested by the paucity (n = 13) of large series (ie >=10 patients) reported.
The infrequency of AOSD has therefore contributed to the lack of reliable epidemiologic
data regarding the incidence or prevalence in an adult population. The frequency of AOSD
may therefore be inferred from rates observed in a juvenile population, and data drawn
from large clinical series of AOSD and population based reports regarding fever of unknown
origin (FUO).
| *Author/year | *No. (%) | *Time Period (yrs) | *Patients Identified From |
| Bywaters 1971 | 14 | 20 | Rheumatology unit survey |
| Goldman 1980 | 13 | 4 | Rheumatology unit survey |
| Del Paine 1983 | 7 | 9 | Rheumatology unit survey |
| Cush 1985 | 21 | 15 | Rheumatology unit survey |
| Bujak 1973 | 10(5%) | 11 | 200 FUO patients |
| Aduan 1979 | 21 (6%) | 15 | 347 FUO patients |
| Larson 1982 | 5 (5%) | 10 | 109 FUO patients |
| Kazanjian 1992 | 5 (6%) | 6 | 86 Community FUO patients |
| Knockaert 1993 | 4(9%) | 9 | 45 "Episodic FUO" patients |
Table 4. Frequency of AOSD
Studies from the University of
Pittsburgh identified 21 AOSD patients seen at once center over a 15 year period (Cush
1986). Bywaters has reported a similar prevalence by reporting 14 patients from two
hospitals over a twenty year period. Bujak et al identified 10 patients from 200
consecutive FUO patients under evaluation at the NIH. Additional data can be ascertain
from the frequency of AOSD among patients with fever of unknown origin (FUO). In 1973
Bujak et al reported a 5% prevalence of AOSD among 200 FUO patients referred to the NIH.
Larkin and Petersdorf (1982) also demonstrated a 4% prevalence of AOSD among 109 patients
reviewed between 1970 and 1980. In most large series, AOSD has a 59% frequency and been
noted to be the most common rheumatic cause of FUO (Aduan 1979, Larkin 1982, Kazanjian
1992). From these data and unofficial surveys of rheumatology units around the USA, a
reasonable estimate would suggest an annual incidence rate of 13 new AOSD patients/year
at most major medical referral centers.
| *FEATURE | *(+)/TOTAL | *PERCENT |
| Male | 173 | |
| Female | 189 | |
| JRA Hx | 39/258 | 15.1% |
| Quotidian T > 39F | 301/320 | 94.1% |
| Fever T > 39F | 346/349 | 99.1% |
| Still's Rash | 311/358 | 86.9% |
| Rash | 259/281 | 92.2% |
| Alopecia | 20/136 | 14.7% |
| Arthralgia | 295 | 99.7% |
| Arthritis | 314/338 | 92.9% |
| Destr/Erosive Dz | 55/253 | 21.7% |
| TMJ Pain | 24/129 | 18.6% |
| Cervical Pain | 24/52 | 46.2% |
| Myalgia | 175/235 | 74.5% |
| Wt. Loss > 10% | 83/127 | 65.4% |
| Sore Throat | 190/268 | 70.9% |
| Lymphadenopathy | 193/300 | 64.3% |
| Splenomegaly | 139/327 | 42.5% |
| Hepatomegaly | 91/228 | 39.9% |
| Hepatotoxicity | 12/176 | 6.8% |
| Pleuritis | 111/311 | 35.7% |
| Pericarditis | 90/301 | 29.9% |
| Pneumonitis | 50/229 | 21.8% |
| Abdominal Pain | 46/153 | 30.1% |
| Pos. RF | 17/341 | 5.0% |
| Pos. ANA | 7/334 | 2.1% |
| Elevated ESR | 288/291 | 99.0% |
| WBC >=10.0 | 282/305 | 92.5% |
| WBC >=15.0 | 173/244 | 70.9% |
| Thrombocytosis | 44/97 | 45.4% |
| Hct < 35% | 181/249 | 72.7% |
| Pos. Coombs | 5/74 | 6.8% |
| Hemolysis | 2170 | 2.9% |
| Albumin <3.5gms | 96/126 | 76.2% |
| Elevated LFT's | 186/265 | 70.2% |
| Proteinuria | 28/82 | 34.1% |
| Carpal Ankylosis | 100/233 | 42.9% |
| Cervical Ankylosis | 18/148 | 12.2% |
| Tarsal Ankylosis | 16/84 | 19.0% |
| Sacroiliitis | 10/110 | 9.1% |
Table 5. Clinical Manifestations of 382 AOSD Patients from World Literature
Clinical Manifestations
AOSD is defined as a systemic inflammatory disorder of unknown etiology and pathogenesis that typically afflicts young adults. Characteristic manifestations include quotidian fevers, evanescent rash, pharyngitis, arthralgias, arthritis, hepatosplenomegaly, polyserositis, leukocytosis, and negative serological tests for antinuclear antibodies (ANA) and rheumatoid factor (RF) (Table 5). Its clinical course is marked by systemic exacerbations and/or chronic arthritis, frequently with diseasefree intervals.
This multisystem inflammatory disease may manifest
differently throughout the course of disease. Prominent systemic features are
characteristic of AOSD early in the process, while articular manifestations may dominate
the picture with chronicity. Nonetheless, all patients have a near textbook account of
systemiconset JA at the onset. Most patients will relate similar symptomatology with
striking reproducibility from day to day. These symptoms evolve in a predictable daily
fashion. This point is well illustrated by the wife of a 53 year old AOSD patients who
kept a diary account of her husbands illness (Figure 2).
Illness: Unknown crippling malady
Symptoms: Begins with an intense sore throat. Muscle weakness and soreness. Joints
red and swollen with soreness. Itchy rash. Hearing loss. Can't eat. Alternatively feverish
and shaking with cold. Practically immobile.
History: First had illness April 23rd and went to the hospital a week later.
Chances of survival looked bad. Diagnosis was rheumatic fever. After the first week, it
was decided that this was a wrong diagnosis. Tests of every sort were made all proved
negative. He was sent home May 19th, though he never felt good. His fever
stayed up around 103°F. June was bad for him. By July he was working full time again.
Clearly is woman recorded several key features of this
disease. This patient manifested with a characteristic rapid and severe onset of sore
throat, cyclic fevers, rash and diffuse musculoskeletal manifestations. He looked gravely
ill during the onset and underwent a lengthy and extensive search for an occult process.
Response to antiiflammatory therapies sometimes takes weeks to months. This case also
illustrates that the duration of disease and the potential for rapid recovery is
unpredictable.
Figure 2. Letter from wife of patient DM:
Sex and Age. Contrary to early disparate reports, males (48%) and females (52%) are equally affected. As this disease appears to be the adult counterpart of systemiconset JA, the vast majority of AOSD patients are young adults with nearly 75% having their disease onset prior to age 35 (Figure 3). Although inclined to affect the young, AOSD has been reported in all age groups. Wouters et al (1986) noted that 26% of his 42 patients had a disease onset after the 35 years of ages. Less than 10% of patients will have their onset after age 50. Several patients have been reported with onset of AOSD in their 8th decade of life (Steffe 1983, Koga 1992, Uson 1993, Wouters 1986). This small subset of very late onset Adult Still's disease are often difficult to diagnose, primarily because of age-related comorbid conditions, atypical cutaneous features and low grade fevers, rather than the spiking, characteristic fever of AOSD.
Disease Onset. In the vast majority of patients, the onset of disease is heralded by a sore throat and other constitutional manifestations reminiscent of a viral syndrome. A prodromal sore throat is seen in over 70% of AOSD patients, seems to associate with onset or flare of disease activity. This nonexudative pharyngitis usually lasts days rather than weeks, is without microbacteriological evidence for infection and is unresponsive to antibiotics. It is unknown whether this represents mucosal infection or is a manifestation of AOSD related inflammation (Elkon 1982) is unknown. At the onset, constitutional features may be prominent and are often bothersome to the patient. These include severe myalgias or arthralgias, fatigue, anorexia, nausea and rapid weight loss. Weight loss is seen in 50-60% of patients and may be dramatic and rapid. Numerous patients have reported weight loss of 10-20 kg in the first month of disease activity. Overall, the constitutional manifestations will parallel the severity of systemic activity. Myalgias, fatigue, rash and serositis often show a diurnal rhythm and seem to worsen with the cyclical paroxysms of fever.
AOSD - AGE/SEX
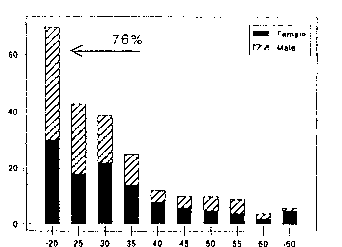
Figure 3. Age/Sex at Onset
In most patients, the characteristic fevers and evanescent rash commence within 2-3 weeks of the sore throat. In a majority of individuals arthritis will manifest contemporaneously with fever and rash or shortly thereafter. However, few case reports have appeared in which an articular presentation antedates the fever and rash by up to 6 months. Del Paine and Leek (1983) point out that 5/7 patients had fever before arthritis and only 1/7 patients had arthritis before the onset of fever. There are no reliable parameters in onset period useful in predicting the severity of disease, clinical course or therapeutic response.
Quotidian Fever. Over 99 % of patients with AOSD manifest with fever > 39 0 C at sometime during the course of their disease and greater than 94% will demonstrate the high spiking, quotidian pattern. Fever is highest during the onset of AOSD and in the younger age groups. Low grade fever and atypical fever patterns are sometimes encountered in older patients (> 35 years) and sometimes late in the course of active disease, especially if articular manifestations dominate the clinical picture. The fever pattern in AOSD is characteristically quotidian (once/day) and sometimes double-quotidian (twice daily) in pattern. A quotidian pattern produces daily febrile spike(s) and later returns to a normal baseline. A remittent pattern is characterized by spikes and an elevated baseline temperature. Bujak et al (1973) reported that 9/10 patients manifested a single quotidian pattern, 3/10 had a double quotidian pattern and 2/10 patients had a remittent pattern. With the initiation of anti-inflammatory therapy or clinical improvement fever patterns may change (ie, a remittive fever may become quotidian or a double quotidian fever may become single quotidian). The highest recorded temperature to date was 107.60F and nearly two-thirds of all patients will have peak temperatures of greater than 400C (Figure 4).
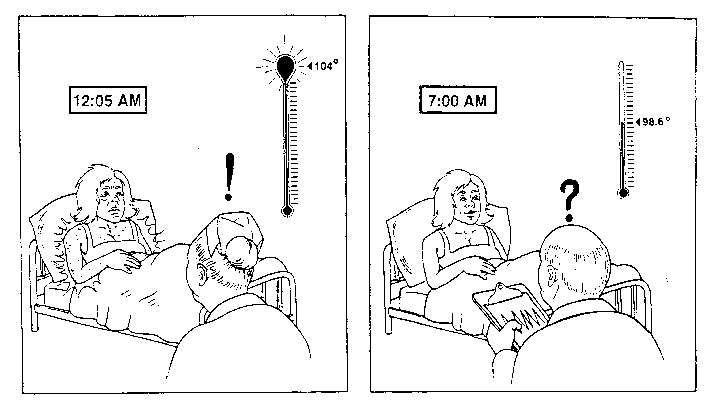
The febrile paroxysms are cyclic and tend to recur every 24, or sometimes every 12, hours. In 1897, Still noted the "pyrexial attacks show a curious regularity in their recurrence". Bywaters has noted that fever is "characteristically very high in the evening, returning to normal by morning.." (Bywaters 1967). The chronology of febrile paroxysms are quite similar from patient to patient. Paroxysms are heralded by shaking chills, followed by 2-4 hours of high fever (> 104°F), and ending with defervescence and drenching sweats. A majority of AOSD Patients will note the onset of fever usually late at night (between 10 PM-2 AM). It has also been described in the late morning (11-12 AM) or late afternoon (4-6 PM). More importantly, individual patients demonstrate a striking tendency to reproduce the fever at the same time of day, each day, until appropriate anti-inflammatory therapy is begun. As such, AOSD patients often appear normal during early morning physician rounds and tend to "flare" late at night when rash and fever may go unnoticed or undocumented (Cover illustration). Febrile spikes are often accompanied by contemporaneous exacerbation of other systemic manifestations (ie, rash, myalgias, arthralgias, fatigue, serositis, headache or nausea). The degree and frequency of fever correlates best with constitutional manifestations, less so with other systemic complaints (serositis, RES, leukocytosis, ESR), and shows little relationship to articular manifestations.
AOSD - FEVER
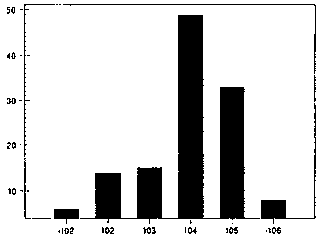
Figure 4. Tmax ln AOSD
Evanescent Rash. The classic, evanescent rash of Still's disease was first noted by Boldero in 1933 and is referred to as a "Still's rash" or "rheumatoid rash" despite the absence of an association with adult seropositive rheumatoid arthritis. While 92% of all patients demonstrate some cutaneous manifestation during their illness, the more specific Still's rash is seen in 86% of patients with AOSD. This rash displays a characteristic periodicity, appearance and location (Table 6) (Isdale 1956). Evanescent by definition, the rash frequently appears during febrile attacks and may last for several hours following defervescence. In general, the Still's rash lasts hours and changes daily, although in some patients, the duration of rash correlates well with the degree of systemic activity and may last for days without change. It is typically salmon-colored (infrequently erythematous), maculopapular and may be confluent or show areas of central clearing. It is usually found on the extremities (extensor surfaces), trunk, neck, and rarely manifests on the face. Two characteristic and common findings are Koebner phenomena and dermatographism. Koebnerization refers to the reproduction of characteristic isomorphic lesions at sites of earlier physical trauma. Dermatographism is an exaggerated cutaneous urticarial response to cutaneous stimuli (ie, the scratch test). While some claim this rash is typically nonpruritic, I have found that up to 35% of patients have complained of pruritis at sometime during their course and that urticaria is not uncommon. Seldom is pruritis a prominent manifestation and if so, should raise suspicions of alternative diagnoses, such as primary biliary cirrhosis or urticarial vasculitis. Kaur et al (1994) has recently described another cutaneous manifestation of AOSD, namely persistent dermal plaques which have the same distribution but, are usually erythematous. Cutaneous anergy is also common in the face of active disease.
- Characteristically evanescent (worse with fever)
- Salmon-pink, faint erythema.
- Maculopapular on trunk, neck; & extremities
- Dermatographism
- Koebner phenomenon
- Uncommon in AOSD: pruritus, urticaria, dermal plaques, facial rash, alopecia, erythema, nodosum, Raynauds
Table 6. Rheumaloďd or Stills rash.
Atypical cutaneous manifestations include alopecia, raynauds phenomenon, and petechial, malar, circumoral or erythema nodosum-like lesions. Cutaneous manifestations of AOSD are most prominent early in the disease and tend to decline with time, evanescent rashes are uncommon in those with more than 10 years of disease.
Pathological examination of lesional skin shows a nonspecific chronic inflammatory picture with a perivascular mononuclear, and seldom polymorphonuclear, infiltrate, vascular dilatation and dermal edema. Serial skin biopsies done by Ridgway (1982) showed episodic deposition of circulating immune complexes. In all instances skin biopsies and immunofluorescent studies have been nondiagnostic.
Articular Manifestations. The presence of arthritis completes the triad of AOSD, however its presence and extent is less evident at the onset than during the disease course. As stated earlier, it is rare that AOSD will present with arthritis prior to the onset of other systemic and extraarticular manifestations. Morning stiffness, myalgias and arthralgias dominate the early clinical picture. Tenosynovitis is also common early on and is often overlooked or mistaken for arthritis or arthralgias (Table 7).
| COMMON | UNCOMMON |
| Myalgias | Tenosynovitis |
| Arthralgias | Periostitis |
| Fleeting Arthritis | Tarsal ankylosis |
| Chronic Polyarthritis | Cervical ankylosis |
| Syn. Fluid WBC= 3-40K | Myositis |
| Carpal Ankylosis | Micrognathia |
| Erosive Hip Dz | Rhabdomyolysis |
| HLA-DR4(+) | DIP capsular calcification |
Table 7 Musculoskeletal Features of AOSD
During the first 6 mos. the onset of polyarthritis is expected in > 90% of patients and may involve large and small articulations. Affected joints most commonly include (in decreasing frequency): the knees, wrists, ankle, elbow, shoulder, PlPs, DlPs, TMJ and cervical spine. Early on. synovitis may be fleeting, migratory or additive (Figure 5). However, with chronicity synovitis, predominantly involves the wrist, with a less commonly the tarsal, cervical, PIP and DIP joints. Neck pain is seen in nearly half of patients at sometime in their disease and is either due to cervical myalgia or arthritis. Several authors have noted the late development of Heberdens-like lesions at the PIP joints in patients (Cush 1985, Wouters -1986). Sacroiliac involvement is uncommon, but has been reported by Bywaters (1971) and Goldman et al (1980). Although both symmetric and asymmetric polyor oligoarthritis is commonly observed, chronic monarthritis is decidedly rare and should suggest other diagnoses.
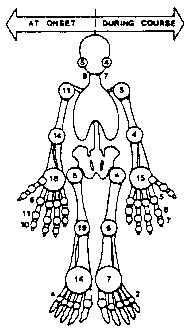
Figure 5 Arthritis in AOSD
The association of AOSD with bony ankylosis of carpal and carpometacarpal articulations first noted by Medsger and Christy in 1976 has since been confirmed by others (Wouters, Pouchot, Zajaczek-Grabowaka). The incidence of carpal carpornetacarpal ankylosis approaches 50% for most clinical series and is also prevalent among systemic-onset JA patients (Talesnik). Bony ankylosis tends to be a symmetric process occurring as a consequence of active synovitis. Once ankylosis is complete, the wrist is a painless, irreversibly fused articulation. The incidence of bony ankylosis is somewhat greater if one examines the tarsus and cervical spine. Ankylosis at these sites is further radiographic evidence favoring a diagnosis of AOSD. Ankylosed articulations are rarely evident on initial evaluation or early in the disease, but evolve quickly (ie. 18-36 mos.) in the face of active articular disease. Whether the presence of ankylosis portends a graver articular course is debatable, however carpal ankylosis is more common among those with a chronic articular course (Cush 1985).
The risk for erosive and destructive polyarthritis is significant, especially in those with a chronic polyarticular course and in those with hip involvement (Wouters 1986, Cush 1985). In the Pittsburgh study, we found an 19% incidence of disability (ARA functional class III/IV disease) among our AOSD patients, with an even greater risk (40%) for those with a polyarticular onset and course (Cush 1985). Nearly 20-25% of all patients will progress to class III/V disease because of early erosive and late secondary degenerative arthritis. Erosive disease has its greatest impact on weight bearing joints. The most common form of joint surgery is hip joint replacement.
Arthrocentesis frequently reveals type II inflammatory synovial fluid. Leukocyte counts usually range from 3.0-40.0 x 103 WBC/mm3, often with a polymorphonuclear predominance. Several reports have noted a decrease in synovial fluid complement levels. Numerous synovial biopsies have consistently shown a chronic synovitis, with proliferation of synovial cells and infiltration of lymphocytes and plasma cells. Electron micrographs have demonstrated type B synovial cell hypertrophy.
Muscle Involvement. Generalized myalgias are seen in 75% of AOSD patients and is frequently a prominent complaint at the onset of disease. Occasionally, mild-moderate elevations of muscle enzyme have been reported, as have poorly documented cases of myopathy and abnormal electromyograms (Esdaile 1979, Bujak 1973). Nonetheless, overt myopathy or myositis has seldom been reported (Moreno-Alvarez 1993, Samuels 1989, Schwarzberg 1982). Similarly, myocardial involvement has been rarely noted in both children and adults with Still's disease (Bank 1985, Hosaka 1992, Sachs 1990, Ward 1988). One should not overlook the possibility of myositis or myocarditis if CPK elevations are present. The distinction between AOSD and dermatomyositis may be difficult (Moreno-Alvarez 1993), but is likely to be determined by the degree of muscle inflammation and the character of the cutaneous lesions. Rare report of rhabdomyolysis (Samuels) and diaphragmatic weakness due to myositis (Braidy 1984) have appeared in the literature.
Reticuloendothelial Disease. Lymphadenopathy, hepatornegaly (with or without hepatic dysfunction), and/or splenomegaly is very common early in the disease and reflects tissue infiltration with inflammatory cells and heightened immunologic activity within the reticuloendothelial system (RES).
Palpable or radiographic demonstration of splenomegaly is seen in 42% of individuals and is most likely the result of hyposplenism and passive congestion. Hyposplenism and splenic sequestration of red blood cells may account for the rapid anemia and splenomegaly often seen in the acute stages of AOSD. Elkon et al (1982) utilized radiolabeled RBC's in 9 AOSD patients to gauge their splenic function. Both active patients and those in remission demonstrated impaired clearance of RBC's. Other investigators have used this method of testing splenic function and have shown an inverse relationship between the presence of circulating immune complexes and splenic clearance of RBCs. These authors postulated that hyposplenism may predispose AOSD or alternatively, splenic function may be impaired as a result of AOSD. Pathological analysis provided by splenectomy during exploratory laparotomies have shown sinus hyperplasia, histiocytosis and slight infiltration of leukocytes (Esdaile, Bujak, Medsger).
While nearly 40% of patients are found to have hepatomegaly, nearly 70% demonstrate abnormalities of hepatic enzymes at some time during their illness. Liver biopsies done in the face of active systemic disease have demonstrated periportal mononuclear infiltrates (lymphocytes, plasma cells) and Kupffer cell hyperplasia. Lobular inflammation, focal hepatocellular degeneration, and periportal fibrosis have also been infrequently noted by some authors. Tender hepatomegaly well with abnormalities of hepatic enzymes, which show equal percentage of hepatocellular and cholestatic inflammatory changes. Further evidence of hepatic dysfunction is provided by modest elevations in the prothrombin time and often impressive hypoalbuminemia. Hypoalbuminemia may be rapid and profound and is seen in 76% of AOSD patients. Elevation of the serum bilirubin is uncommon, but when present may suggest active hemolysis, salicylate-induced hepatotoxicity, or severe hepatocellular damage and risk of hepatic failure (Reginato 1987). Chronic liver disease has been reported in one -individual, with the authors stressing the discordance between the hepatic dysfunction and other features of AOSD activity. This is in contrast to most individuals in whom elevations of hepatic enzymes often subsides with the initiation of anti-inflammatory therapy. These changes of inflammatory hepatitis are sequelae to systemic inflammation and heightened cytokine activity (ie, IL-6, IL-1).
A relationship between hepatic dysfunction and salicylate-induced hepatotoxicity in AOSD has been suggested, but is probably overstated. The incidence of salicylate hepatotoxicity in juveniles with Still's disease is far greater than that seen in this adult population and may reflect the functional immaturity of the liver in a juvenile population. The risk for hepatotoxicity in this population is associated with high doses of salicylates therapy (90-100 mg/kg) and the secondary rise in unbound, pharmacologically active ASA with the disease-related decline in serum albumin. Rich and Johnson reviewed ASA hepatotoxicity in the juvenile population and suggested that ASA hepatitis 1) was dose related and reversible; 2) manifested identical pathology and enzyme elevations, as AOSD hepatitis; and 3) that eosinophilia preceded the rise of enzymes and the subsequent fall in eosinophil count coincided with maximal hepatic dysfunction (Rich 1973). Our review discloses 12 well-documented cases of salicylate hepatitis in AOSD. Esdaile et al (1979) demonstrated that 60% of salicylate treated patients improved their hepatic abnormalities and stated that "..in some patients ASA may be acting to unmask underlying disease related abnormalities". Acute liver failure and chronic liver disease have been sporadically reported (Tesser, Esdaile, Reginato).
Lvmphadenopathy is seen in 65% of AOSD patients. Despite the frequency of this finding, lymphadenopathy is seldom the dominant presenting feature. Lymphadenopathy most frequently manifests as generalized mild to moderate nodal enlargement of nontender lymph nodes located in the cervical, axillary, epitrochlear, or inguinal regions. Mesenteric, para-aortic and hilar nodes may be discovered during diagnostic imaging. Tenderness or focal enlargement of a solitary node or chain should suggest either infectious or neoplastic etiologies rather than AOSD (Table 8). Lymphangiograms are of no value and lymph node biopsies yield nonspecific, reactive hyperplasia or lymphadenitis, sometimes accompanied by histiocytic infiltration.
- Focal, tender, or rapidly changing adenopothy
- Adenopathy in AOSD patients. > 50 yrs.
- When clinical/lab features conflict with Dx of AOSD
- Peripheral smear - shows many myeloid precursors of atypical lymphocites
Table 8. When to Consider Lymph Node Biopsy in AOSD
On occasion, lymph nodes removed from patients with established or presumed AOSD show atypical changes that may question the diagnosis of AOSD. At least 5 cases of Kikuchi's syndrome or necrotizing lymphadenitis, have been described (Ohta 1988, Lyberatos 1990) in patients with established AOSD. Kikuchi's syndrome is a benign disorder, often associated with viral infection, and may manifest as fever, tender lymphadenopathy, hepatomegaly, leukopenia and elevated ESR (Ohta 1988). Because of similar features Ohta et al has suggested that it is plausible that many of the patients reported with Kikuchi's syndrome may have AOSD also. Alternatively, necrotizing lymphadenitis may be a milder more localized form of the more systemic RES involvement seen in AOSD (Ohta 1990). Histologically, Kikuchi's syndrome must be distinguished from malignant lymphoma, angioimmunoblastic lymphadenopathy, etc.
Valente et al (1989) reviewed 8 lymph node biopsies from 97 AOSD patients seen at the Mayo Clinic and found distinctive pathology in 7/8 AOSD patients. These biopsies demonstrated an intense, somewhat atypical, paracortical hyperplasia with nodal effacement and scattered atypical immunoblasts. Although the original pathologic interpretations included malignancy (3 cases), mixed lymphoma (1 case) and angioimmunoblastic lymphadenopathy (2 cases), prolonged follow-up confirmed the diagnosis of AOSD. Several other authors have questioned the lymphomatous appearance of AOSD lymph node biopsies (Quaini 1991, Reichert 1992), while others have documented the occurrence of lymphoma in patients with established AOSD (Trotta 1993, Kawasaki 1995). Trotta et al described a case of B cell lymphoma (associated with a trisomy 12 karyotype) arising 2 years after typical AOSD without lymphadenopathy (Trotta 1993). Biopsy was prompted by occurrence of persistent lymphadenopathy and the development of hypogammaglobulinemia, paraproteinemia and hyperferritinemia. In contrast, Reichert (1992) has reported a case of AOSD wherein the lymph node histology was interpreted as "malignant T cell lymphoma". Nonetheless, the patient responded well to minimal anti-inflammatory therapy only and showed no evidence of lymphoma after 3 years of therapy. These authors appropriately point out that the clinician should be aware that "lymph node histology in AOSD may mimic malignant lymphoma".
Serositis. Pleuritis (40%) and pericarditis (30%) are quite common when detected by pleuritic chest pain, auscultatory rub, or the demonstration of pleural or pericardial fluid by abnormal chest radiographs or echocardiogram. In some, the pain of serositis may be the presenting complaint, while in others it may be an asymptomatic finding. Pleural effusions are usually bilateral, seldom large enough to be symptomatic, and rarely produce pleural thickening. Thoracentesis often yields bloody, exudative effusions with white blood cell counts ranging from 3-20 x 103/mm3 with a polymorphonuclear predominance. Pleural biopsies have not yielded diagnostic information showing only acute and chronic inflammation.
Pericarditis is the more worrisome finding as some AOSD patients have presented as acute cardiac tamponade and have required emergency pericardiocentesis or pericardial stripping (Alukal, Jamieson, Strumpf, Drouot, Esdaile, Desablens). While pericardial tamponade is quite rare, pericarditis is seen in 30% of AOSD patients. Clinically, such patients present with acute sharp chest pains, with dyspnea and partial positional relief. Documented cases have responded well to drainage and high dose corticosteroids. Pericardial fluid is frequently bloody and usually exudative with WBC counts ranging from 1.2-26,000 (PMN predominance). Myocarditis is rarely seen in AOSD, but tends to occur in patients with very active systemic disease and evidence of pericarditis and/or pleuritis.
Pneumonitis is found in over 20% of patients and often presents a diagnostic challenge. These individuals often appear septic with complaints of fever, dry cough, dyspnea and are found to have pulmonary infiltrates that are unresponsive to anti-infective therapy. Infiltrates tend to be bilateral more commonly than unilateral, alveolar or interstitial in pattern and responds well to anti-inflammatory therapy with steroids. Several authors have noted restrictive changes on spirometry and few progress to chronic respiratory insufficiency as a result of aggressive parenchymal inflammation (Pedersen 1991, Corbett 1983, Cantor 1987, Vaan hoeyweghen 1993). Braidy (1984) has demonstrated that diaphragmatic weakness from myositis may contribute to the pulmonary pathology noted.
Abdominal pain is seen in 30% of patient, primarily during the onset phase. Abdominal pain is often diffuse and may be accompanied by nausea, vomiting and sometimes diarrhea, but seldom do these features dominate the clinical picture. Abdominal pain has been ascribed to serous peritonitis (Pollet 1990, Bujak 1973), mesenteric adenitis, hepatic or splenic distention, ileus or small bowel obstruction.
Miscellaneous Features and Uncommon Associations. Renal disease is uncommon, yet many patients will manifest minor and transient proteinuria (34%) during periods of febrile paroxysms. Chronic or substantial proteinuria is uncommon and likely to be secondary to amyloidosis. Amyloidosis has been reported in a handful of patients and should be suspected with persistent hematuria and proteinuria. While most cases have been reported from Europe (Wendling, Hory), several AOSD patients from the United States have been reported (Harrington 1981, Elkon 1982, Larson 1985, Medsger 1976, Terkeltaub 1981). CNS disease is also rare and may manifest as sensorineural hearing loss, mental status changes, aseptic meningitis, meningoencephalitis, peripheral neuropathy, ulnar tunnel syndrome or orbital tenosynovitis (Browns syndrome) or inflammatory orbital pseudotumors. Other uncommon associations include leukopenia, mild eosinophilia, disseminated intravascular coagulation (DIC), Sjogrens syndrome, Raynauds, nodules, uveitis, episcleritis, retinal hemorrhages or exudates, psoriasis, autoimmune thyroid disease, glomerulonephritis, nephrolithiasis, hemophagocytic syndrome and Takayasu's arteritis.
Several authors have suggested an association between AOSD and pregnancy. However, the the lack of a consensus (despite the number of patients reported) and the high percentage of young women among AOSD patients suggests that this an association is coincidental rather than causal.
Laboratory and Radiographic Abnormalities
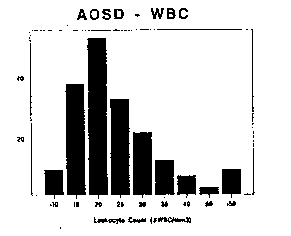
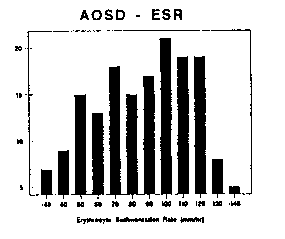
Figure 6 WBC counts in AOSD Figure 7 ESD in AOSD
In parallel with systemic inflammation, laboratory investigations are notable for the consistent absence of antinuclear antibodies and rheumatoid factor, but the impressive degree of neutrophilic leukocytosis (92%) and elevated sedimentation rates (Table 5, Figures 6 and 7). The simultaneous occurrence of both may prove to be a valuable feature in the diagnosis of AOSD (Cush 1984). Leukocyte counts generally range between 12,500-40,000 cells/mm3, with the highest recorded to be 69,000. Nonetheless, nearly 20% of patients may demonstrate marginal WBC elevations (10-15K), in which case the identification of other characteristic features will be required to affirm the diagnosis of AOSD. Acute phase reactants are markedly elevated in AOSD, including the ESR, C-reactive protein (CRP), serum amyloid A (SAA), thrombocytosis and serum ferritin. Over 90% of AOSD patients have an ESR > 50 mm/hr and 50% have and ESR > 90 mm/hr. Extreme leukocytosis or leukopenia are rare in AOSD (Scopelitis) and should raise the possibility of an occult leukemia or lymphoma (Trotta 1993, Sugawara 1993).
During periods of active systemic
disease red blood cell counts may drop precipitously and in parallel with the weight loss
and/or serum albumin - all reflecting systemic inflammatory activity. Aplastic anemia has
been reported, presumably as a consequence of therapy (Medsger 1976, Kahl 1988). Rare
reports of hemolytic anemia and/or DIC should question the diagnosis of AOSD or the
possibility of an associated hemophagocytic syndrome (Coffernils 1992, McPeake 1993).
Elevations in the hepatic enzymes
frequently occur (70%) as a manifestation of inflammatory hepatitis, rather than the rare
ASA or NSAIDinduced hepatotoxicity. Both cholestatic and hepatocellular patterns of
enzyme elevations have been observed. Depressed serum albumin levels between 2.0 and 3.5
mg% is seen in over 75% of patients. Other reported laboratory abnormalities include:
eosinophilia; hypergammaglobulinernia (50% of pts); IgA deficiency (5 cases); circulating
immune complexes (2050% of pts); and elevated ASO titers in onethird of patients.
Serologic tests for complement, HLAB27, cold agglutinins, serum immunoelectrophoresis,
and thyroid studies are consistently normal.
Hyperferritinemia. It has been
suggested that extreme elevations of the acute phase reactant, ferritin, may be of
diagnostic value in assessing patients with AOSD. Hyperferritinemia with values between
4000 30,000 mg/ml have often been reported in association with the onset and/or flare
of disease activity (Ohta 1987, Pelkonen 1986, van Reeth 1994). Levels as high as 250,000
mg/ml have been reported (Coffernils 1992). Ohta has reported on 38 Japanese patients with
active systemic AOSD. Of these, only 7 patients had normal ferritin levels, 26 were
greater than 1000 mg/ml and 20 were greater than 4000 mg/mI (Ohta 1989). In a survey of 20
systemiconset JA patients, Pelkonen found elevated ferritin values in 19/20 JA patients
at the onset of their disease. Values above 1000 ug/l were seen in ~0% of patients, while
only 3/20 patients had values > 4000 ug/l. In contrast, Coffernils et al (1992) has
reported normal levels in 40% of active AOSD patients, 30% with levels between 300 1000
mg/ml and only 30 % with levels above 1000 mg/ml. Ferritin is found in nearly all tissues,
with the highest concentrations in the reticuloendothelial systems (liver, spleen, bone
marrow) and heart. It has been suggested that highest levels are common among patients
with abnormal hepatic enzymes (Ohta), although this has not been universally observed (van
Reeth 1994, Pelkonen 1986, SchwarzEywill 1992). van Reeth et al (1994). has shown by
isoelectric focusing that circulating ferritins were uniformly basic and almost completely
desialylated and that this distinguished active AOSD from inactive AOSD or patients with
other systemic rheumatic disease. The reason underlying the inadequate sialylation of
ferritin in AOSD is unknown, but may contribute to its impaired clearance and accumulation
in the circulation. van Reeths patients with active AOSD demonstrated exceptionally high
mean ferritin values (4519 mg/ml) when compared with inactive AOSD patients (233 mg/ml)
(Figure 8). Multiple reports have shown a close correlation between elevated ferritin and
the high spiking fevers of Still's disease (Pelkonen 1986). Moreover, ferritin levels are
rapidly responsive to effective anti-inflammatory therapy (Pelkonen) (Figure 9).
Ferritin is known to increase in the face of infection, neoplasia and inflammatory states, and is thus an acute phase reactant. Ferritin synthesis and release is strongly influenced by cytokines (Rogers 1990, Tsuji 199 1). GonazalezHernandez et al (1989) has shown that of the 5,600 ferritin levels done at one center over an 8 month period, hyperferritinemia (> 4000 mg/ml) was observed in 37 instances and these were due to polytransfusion states (14 pts), neoplasia (7), AOSD (4), chronic hepatitis (4), and 2 each with hemochromatosis, pancreatitis, sepsis and 2 patients were undiagnosed. Other common causes of raised ferritin levels include chronic leukemia, malignant lymphorna, melanoma, neuroblastorna, germ cell tumors, hepatic necrosis, and rarely rheumatoid arthritis or SLE. The predictive value of increased ferritin levels is only likely to be helpful if greater than 4000 mg/ml. Otherwise, mild-moderate elevations would reflect a nonspecific acute phase response that might accompany a number of common inflammatory febrile disorders.
Hyperferritinernia in AOSD is
unrelated to iron metabolism and is more likely to be a consequence of cytokine induce
augmented synthesis by the RES or hepatocyte damage resulting in increased release.
Lastly, some reports, have suggested that hyperferritinemia is associated with histiocyte
hyperactivity that may lead to the seldom reported association of Still's disease and the
hemophagocytic syndrome (Morris 1991).
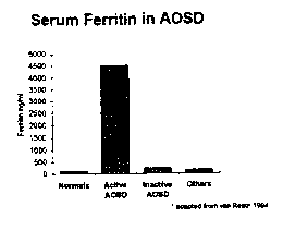
Figure 8 Hyperferritinemia in AOSD
The hemophagocytic syndrome has been
reported to be either familial or associated with viral infections, neoplasms or immune
mediated disorders (McKenna 1981). It is characterized by proliferation of mature
nonneoplastic histiocytes with prominent hemophagocytic activity within the bone marrow,
spleen or lymph nodes. Patients often present with impressive fever, lymphadenopathy,
hepatosplenomegally, and cytopenias. Still's patients have a similar presentation, but are
hardly ever cytopenic. When reported in association with Still's disease, the
hemophagocytosis syndrome has occurred following seroconversion to one Of several viruses,
including: EBV, hepatitis A, varicella and influenza A (Coffernils 1992, McPeake 1993,
Morris 1985). Extremely high ferritin levels are often seen in viral associated
hemophagocytosis syndrome and malignant histiocytosis (Coffernils 1992). The presence of
this complication is associated with very high mortality rates. Disseminated intravascular
coagulation, hemolytic anemia profound cytopenias are often present in those who are
severely affected by this syndrome.
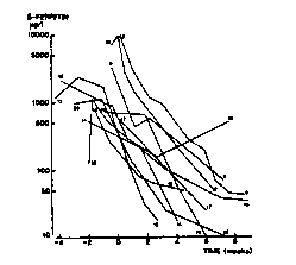
Fig 9 Ferritins response to Steroids in Still's Dz
Carpal ankylosis is a distinctive
feature of AOSD and should be sought for. Nearly half of patients will manifest ankylosis
after several years of disease activity. While carpal ankylosis is common and distinctive
in AOSD, it is uncommonly observed be seen in other familiar rheumatic disorders (ie.,
rheumatoid arthritis, psoriatic arthritis, chronic Reiter's syndrome), thus limiting its
predictive diagnostic value. Nonetheless, the identification of early erosive disease and
osteopenia, followed by late ankylosis in a pericapitate distribution (Figure 10) may have
diagnostic and possibly therapeutic implications. Similarly, a striking tendency for
intertarsal ankylosis and ankylosis of the zygoapophyseal joints of the cervical spine
(GarciaMorteo 1988, Bywaters 1971, Healy) has been reported.
Differential Diagnosis
For those who manifest the AOSD triad
of fever, rash and arthritis, diagnostic considerations Must be tailored to the age and
distinguishing features at presentmost difficult cases involve the differentiation between
AOSD and patients with acute systemic viral syndromesation. Personal experience suggests
that. the , Reiters' syndrome, dermatomyositis, or inflammatory bowel disease. Depending
on the clinical picture, AOSD will need to be distinguished from: infectious etiologies,
mycobacterial infection, subacute bacterial endocarditis, occult abscesses, leptospirosis,
brucellosis, malaria, granulomatous hepatitis, sarcoid, Whipples' disease. acute rheumatic
fever, Sweets syndrome, serum sickness, hypocomplementemic urticarial vasculitis,
polyarteritis, lymphoma and leukemia.
Figure 10 Pericapitate pattern of ankylosis in AOSD
Bywaters has suggested up to half of
juvenile arthritis patients classified as "probable Still's disease" will go on
to develop either psoriatic arthritis, ankylosing spondylitis, 1BD arthritis, rheumatic
fever, SLE, dermatomyositis, scleroderma, leukemia, reticulosarcoma, or neuroblastoma
(Bywaters 1967). Other diagnoses that have commonly been misconstrued with AOSD include:
EBV lymphadenitis, acute leukemia, psittacosis, myelodysplastic syndrome and lymphoma.
Diagnosis
In decades past, AOSD patients were
subjected to numerous invasive and noninvasive investigations in search of an occult
infective or neoplastic diagnosis. In due course it has become apparent that this syndrome
lacks a diagnostic serologic test or histopathology and thus, today AOSD remains a
clinical diagnosis of exclusion.
With greater awareness of this
syndrome, AOSD is now being considered earlier in the course of evaluation patients with
fever, dermatitis and arthritis. Whereas prior to 1980, knowledge of AOSD was the major
obstacle to diagnosis, henceforth the primary challenge will rest with the ability to make
an early and correct diagnosis. Requisite steps should include a comprehensive,
noninvasive workup, documentation of fever pattern, and patient observation over a minimum
of at least 6 weeks prior to the diagnosis of possible AOSD. A probable diagnosis will
required an extended period (ie, 6 months) of followup and reassessment. Guidelines
published for the evaluation of FUO are applicable to AOSD patients and will minimize the
cost and invasiveness of diagnostic testing (Smith 1986, Kazanjian 1992).
Diagnostic criteria have been developed by various investigators (Table 9) and each have been tested for sensitivity and specificity in both AOSD patients and FUO controls (Yamaguchi 1992). Whereas the criteria of Cush and Yamaguchi both have specificities greater than 92%, the sensitivity of Yamaguchi is superior to Cush (96% vs. 80%, respectively). Nonetheless, these criteria may prove useful in the study of other AOSD patients or in the evaluation of individual patients with presumed AOSD.
Reginato et al 1987
Definite AOSD: all 4 major Major Manifestations Minor Manifestations Persist/intermittent fever
Serositis |
Yamaguchi et al 1992 AOSD Total of > 5 criteria (including 2 major)
Major Criteria Minor Criteria Fever > 39°C
Sore throat
|
Goldman et al 1980 * High spiking fever
|
Cush 1984 Definite AOSD: > 10 points + 6
months observation Major Criteria (2 points) Minor
Criteria (1 point) |
Table 9 : Proposed Diagnostic Criteria for Adult-Onset Still's Disease
Therapy of AOSD
Therapy is directed at controlling inflammatory symptoms and signs without exposing the
patient to unacceptable toxicity. Historically, the juvenile literature tells us that high
dose salicylate (ASA) is the drug of choice. Responses to ASA similar to that seen in
acute rheumatic fever are likewise thought to be of diagnostic import. Nevertheless, the
risk of Reyes syndrome and the incidence of ASAinduced hepatotoxicity limits the utility
of ASA in the systemiconset JA population. In adults, ASA is better tolerated but, is
only marginally efficacious, with less than one third of AOSD patients responding during
the acute phase of their illness. Nonsteroidal anti-inflammatory drugs (NSAID) have been
used for over 20 years and have been shown to be effective in up to 60% of AOSD patients
(Table 10). Indomethacin (1 2 mg/kg/day) has been the most utilized NSAID, although all
other Nasals may be equally effective when used at comparable anti-inflammatory doses. The
sustained release form of Indomethacin is preferred by many patients because of once or
twice daily dosing (ie. IndocinSR 75 mg. bid). The practice of adding ASA to high dose
NSAID therapy for a presumed additive or synergistic effect, should be discouraged because
of the unacceptable toxicity without apparent benefit.
A majority of patients will require daily corticosteroids to control systemic features of AOSD. Unfortunately, low doses of prednisone (ie., < 20 mg/day) and alternate day therapy have proven ineffective in most patients. Once the decision is made to start corticosteroids, prednisone should be started at 40-60 mg/day, in divided doses if necessary. Reliance on prednisone will be greatest early in the disease and should diminish over time. Therefore, if the patient is still steroid dependent after 3-6 months, the addition of a steroid sparing therapy with methotrexate (MTX), azathioprine (AZA) or hydroxychloroquine should be considered. Pulse steroids have been utilized and should be reserved for the morbidly ill. Systemic disease activity will require aggressive anti-inflammatory therapy with NSAID, prednisone, MTX or hydroxychloroquine and will not respond to other conventional DMARDs (ie, gold or penicillamine) (Ansell 1991, Cush 1985). Multiple reports have documented the efficacy of low dose weekly MTX in the control of fever.and other systemic features of AOSD. These data suggest that MTX may be used early, as a steroid sparing agent, to control systemic and articular disease and limit the toxicities associated with prolonged high dose corticosteroids therapy (Kraus 1991, Aydintug 1992, Hollingsworth 1989, Samuels 1989).
Chronic articular disease should be managed like rheumatoid arthritis and seems to respond in a similar manner to either gold salts, penicillamine, MTX, AZA, sulfasalazine, etc. Experimental therapies shown to be efficacious in a few patients include: IV gammaglobulins; pulse cyclophosphamide; cyclosporin A; and y-interferon (IFNy) (Silverman 1990, Shaikov 1992, Bjerkhoel 1988, Pernice 1989).
Acute Therapy |
Maintenance Therapy |
|||
| Therapy employed (dose) | Responder | Nonresponder | Responder | Nonresponder |
| ASA only (>
4 gr/d)
28
48
18
2 + indomethacin 11 9 11 0 + prednisone 28 0 - - Indomethacin (2
mg/kg/d)
33
17
17
2 Prednisone only (>= 20mg/d)
71
13
32
3 Gold Salts (IM)
21
9 Remittive Rx MCM, AZA, Plaquenil. SSZ, CTX)
16
7 |
Table 10. Therapeutic Response in 221 AOSD patients
Prognosis
Of 221 therapy patients surveyed above, 33 were in remission off all medications, 107 were still on medication and insufficient data exists for the remaining 81 patients. This is similar to the data gleaned from several large series that conclude that self-limiting disease is uncommon (ie, < 20%) and that intermittent systemic disease or chronic articular disease requiring therapy is to be expected (Cush 1985, Wouters 1986, Pouchot 1991).
The majority of patients with systemic disease only will have self-limiting episodes of less than 1 year and will fair well once the disease abates. However the potential for recurrence and life threatening complications cannot be discounted. Such complications include pericardial tamponade, myocarditis, DI C, hepatic failure, inflammatory pulmonary disease and amyloidosis. For those patients developing chronic articular disease, the long term outcome is largely determined by the extent and duration of articular involvement. Thus, a polyarticular onset and course or the development of hip, disease tends to correlate well with poor functional outcomes after prolonged followup. The presence of HLA DR4 may contribute to a poorer outcomes, while HLA-Bw35 may offer some protection, with a better joint prognosis and a propensity for self-limited disease (Terkeltaub 1981, Wouters 1986).
Etiology
No etiopathogenesis has been acceptably proposed or proven for AOSD. An infectious etiology or trigger has been inferred based upon the prodromal sore throat that heralds the onset or flare of disease. Most clinical manifestations are reminiscent of features seen during self-. limited viral syndromes. Lastly AOSD is occasionally associated with other viral related syndromes, such as the hemophagocytic syndrome and Kikuchi's syndrome Although no etiologic factor has been identified, some have advocated that an infectious, and possibly viral, agent initiates a cascade of the immunological events resulting in the characteristic clinical syndrome of AOSD (Wouters 1988, DeVere-Tyndall 1984, Newkirk 1994). Multiple investigators have demonstrated that persistence of a viral antigens, especially rubella, in patients with JA and AOSD (Newkirk 1994, Chantler 1985). Whether such antigens remain immunogenic and contribute to the pathogenesis of systemic or articular disease is unknown. Reactivity to these ubiquitous microbes may therefore be dependent upon genetic, immunologic and other unidentified host factors. When sought for, microbiologic evidence of coexistent viral infections is found in nearly half of patients. Nonetheless, is unknown whether such identification constitutes causal proof. In many instances these findings are either coincidental or may reflect a nonspecific response to heightened immunologic activity.
- Rubella
- Epstein Barr virus
- Coxackie B4
- Mumps
- Adenovirus
- Echovirus 7
- Cytomegalovirus
- Parainfluenza
- Parvovirus B19
- Staphylocoocus
- Y enterocolitica
- Brucella abortus
- Mycoplasma
- Borrelia burgdorferi
Table 11. Implicated organisms in AOSD
Chantler et al (1985) has demonstrated the persistence of Rubella in the mononuclear cells of nearly one-third of JA patients, including those with systemic-onset JA. Using semi quantitative PCR, Newkirk et al has shown that PBMC from AOSD patients more frequently contain viral genome for rubella than do normal controls (83% vs. 33%). In addition, AOSD patients carry a 4 fold greater amount of viral genome than do normal controls (P=0.03) and that the viral genome is primarily found in B cells and monocytes, but not T cells (Newkirk 1994). These data suggest that AOSD patients may be more susceptible to rubella infection and/or are unable to clear virus from their mononuclear cell population (monocytes and B lymphocytes). Impairment of RES function in AOSD has been suggested by Elkon et al (1982) who has documented hyposplenism and the impaired ability of the RES to clear heat-damaged red blood cells. Lastly, the observation of clinical efficacy using subcutaneous gIFN in the treatment of refractory systemic JA questions whether the anti-viral effect of IFN lead to clinical improvement (Pernice 1989).
AOSD - A Circadian Cytokine Syndrome?
I would like to conclude with the suggestion that AOSD is a cytokine driven disorder and that the curious diurnal variations in fever and other systemic features suggest an Underlying circadian rhythm. Predictable paroxysms of fever have also been noted in malaria wherein the periodicity of tertian or quartan fevers has been linked to the growth cycle of the parasites (Kwiatkowski 1989). Rupture of schizont has been associated with the release of TNFa and other cytokines, capable of acting as endogenous pyrogens, (Kwiatkowski 1993).
Endogenous pyrogens, include a number of well known cytokines, such as interleukin-1 (IL-1), IL-6, TNFa, the interferons, and macrophage inflammatory protein MlPla). The in vivo effects IL-1, IL-6 and TNFa have been well characterized and many these correspond to the distinctive features of AOSD (Table 12). Both IL-1 and IL-6, but not TNFa, have been shown to be produced in a circadian fashion (Zabel 1990, Gudewill 1992, Arvidson 1994).
| Clinical Feature | IL-1 | IL-6 | TNFa |
| Fever >. 39 °C | + | + | + |
| Exanthem | + | 0 | + |
| Myalgies | + | +/- | + |
| Arthralgia/arthritis | + | + | + |
| Weight Loss | + | + | ++ |
| Leukocytosis | + | ++ | + |
| ^ Acute Phase Response | + | ++ | + |
Table 12. Clinical Effects of IL-1, IL-6 and TNFa
It therefore seems likely that IL-6 or IL-1 may act as effector molecules that give rise to many of the features that characterized AOSD. DeBenedetti et al has demonstrated markedly elevated levels of IL-6 in the serum and synovial fluid of patients with systemic-onset JA (SOJA) (DeBenedetti 1991, 1994). Moreover, these levels correlate with the level of thrombocytosis and synovitis. Sequential analysis of IL-6 production showed diurnal variation, with peak levels between 1800 and 2200 hours, with the lowest levels approximating the early morning rise in serum cortisol (Zabel 1990). David et al (1990) has also shown that serum IL-6 levels paralleled the febrile spikes in SOJA. Increases in TNFa followed the fever by 5 hours and IL-1 levels were low during the fever and rose once the fever dissipated.
These data suggest that in Still's disease, the diurnal alterations in fever and inflammatory symptomatology are paralleled by the increased production of IL-6 and possibly, other unidentified pyrogens or cytokines. Factors responsible for the circadian production of cytokines are unknown, but may relate to host susceptibility and immunologic reactivity to viral antigens (ie, rubella) sequestered within monocytes, macrophages and other immunocompetent cells.
REFERENCES
Review Articles, Largo Series and Clinical Outcomes
Bambery P. Thomas RJ. Malhotre HS. Kour U. Shusnurmath SR. DoodharSD. Adult onset Still's disease: clinical experience with 18 patients over 15 years in northern India. Annals of the Rheumatic Diseases. 51(4):529-32, 1992 Apr.
Bujak JS, Aptaker RG, Decker JL, Wolff SM. Juvenile rheumatoid arthritis presenting in the adult a* fever of unknown origin. Medicine 52:431-43, 1973
Bywaters EG. Still's disease in the adult. Annals of the Rheumatic Diseases. 30(2):121-33, 1971 Mar.
Caroit M. Mathieu M. Kahn MF. Seze S da. Still's disease in adults and Wissler-Fanconi syndrome. Revue du Rhumatisme et des Maladies Osteo-Articulaires. 400(1):1-8, 1973 Jan.
Cush JJ. Modager TA Jr. Christy WC. Herbert DC. Cooperstain , LA. Adult-onset Still's disease. Clinical course and outcome. Arthritis & Rheumatism. 30(2):186-94, 1987 Feb.
Del Paine DW, Look JC. Still's arthritis in adults: disease or syndrome? Journal of Rheumatology 10:758-62, 1983
Elkon K8. Hughes GR. Bywaters EG. Ryan PF. Inman RD. Bowiey NB. James MP. Eady RA. Adult-onset StWe disease. Twenty-year followup and further studies of patients with active disease. Arthritis & Rheumatism. 25(6):647-54, 1982 Jun.
Esdaile JM. Tannenbaum H. Lough J. Hawkins D.- Hepatic abnormalities in adult onset Still's disease. Journal of Rheumatology. 6(6):673-9, 1979 Nov-Dec.
Godeou B. Leport C. Parronne C. Salmon-Caron D. Vilde JL. Kahn MF. Long term evolution of adult onset Still's disease seen in an infectious diseases department. Annals of the Rheumatic Diseases. 50(12):968, 1991 Dec.
Goldman JA, Board MR, Casey HL. Acute febrile juvenile rheumatoid arthritis in adults: cause of polyarthritis and favor. Southern Medical Journal 73:555-563, 1980
Harth M. Thompson JM. Ralph ED. Adult-onset Still's disease. Canadian Medical Association Journal. 120020507-10, 1979 Jun 23.
Larson EB. Adult Still's disease. Evolution of a clinical syndrome and diagnosis, treatment, and follow-up of 17 patients. Medicine. 63(2):82-91, 1984 Mar.
Larson EB. Adult Still's disease--recognition of a clinical syndrome and recent experience. Western Journal of Medicine. 142(5):885-71, 1985 May.
Ohta A. YemagucN M. Tsunematsu T. Kasukawa R. Mizushima H. Kastiwagi H. Kashiwazaki S. Tanimoto K. Matsumoto Y. Akizuki M. at al. Adult Still's Disease Research Committee, Japan. Adult Still's disease: a multicenter survey of Japanese patients. Journal of Rheumatology. 17(8):1058-63, 1990 Aug.
Ohta A. Yamogucti M. Kanooke H. Negayoshi T. Hiida M. Adult Still’s disease: review of 228 cases from the literature. Journal of Rheumatology. 14(6):1139-46. 1987 Dec.
Pouchot J. Sampalis JS. Beaudet F. Carette S. Docary F. Salusinsky-Sternbach M. Hill RO. Gutkowski A. Harth M. Myhal D. et al. Adult Still's disease: manifestations, disease course. and outcome in 62 patients. Medicine. 70(2):118-36. 1991 Mar.
Reginato AJ. Schumacher MR Jr. Baker DG. O'Connor CR. Forreiros J. Adult onset Still's disease: experience in 23 patients and literature review with emphasis on organ failure. Seminars in Arthritis & Rheumatisrn. 170):39-57, 1987 Aug.
Sampalis JS, Esdaile JM, Medsger TA Jr, Partridge AJ, Yeadon C, Sonecal JL, Myhel D, Harth M, Gutkowski A, Carette S, Beaudet F, Cush JJ, Frias JF. A controlled study of the long-term prognosis of adult Still's disease. American Journal of Medicine 1995 (in press)
Sampalis JS. Pouchot J. Beaudet F. Carette S. Gutkowski A. Harth M. Myhal D. Sonecal JL. Yeadon C. Williams JI. at al. Arthritis impact measurement scales: reliability of a French version and validity in adult Still's disease. Journal of Rheumatology. 17(12)657-61, 1990 Dec.
Wouters JM. van do Putte LB. Department of Rheumatology, Adult-onset Still’s disease; clinical and laboratory features, treatment and progress of 45 cases. Q J Med 235:1055-65, 1986
Yamaguchi M. Ohte A. Tsunomatsu T. Kasukawa R. Mizushima Y. Kashiwagi H. Kashiwazaki S. Tanimoto K. Matsumoto Y. Ota T. et al. Preliminary criteria for classification of adult Still's disease. Journal of Rheumatology. 19(3):424-30, 1992 Mar.
Juvenile Rheumatoid Arthritis
Bywaters EGL. Heberden oration, 1966. Categorization in medicine: a survey of Still's disease. Ann Rheum Dis 26:185-93, 1987
Cabane J. Michon A. Ziza JM. Bourgeois P. Bietry 0. Godeau P. Kahn MF. Comparison of long term evolution of adult onset and juvenile onset Still's disease, both followed up for more than 10 years. Annals of the Rheumatic Diseases. 49(5):283-5, 1990 May.
Calabro JJ. Other extraarticular manifestation of juvenile rheumatoid arthritis. Arthritis Rheum 20:207-11, 1977
Cassidy JT, Nelson AM. The frequency of juvenile arthitis. J Rheumatol 1988; 15:535-6
Fernandez-Vina M, Fink CW, Stastny P. HLA associations with juvenile arthritis. [Review] Clinical and Experimental Rheumatology 12(2):205-14, 1994
Fink CW. Clinical, Genetic and therapeutic aspects of juvenile arthritis. Clinical Rheumatology in Practice 1(3):100115, 1983
Gupta RC. Milo DM. Still's disease in an adult: A link between juvenile and adult rheumatoid arthritis. American Jour" of the Medical Sciences. 2690037-44, 1975 Jan-Feb.
Hafner R. Truckenbrodt H. Course and prognosis of systemic juvenile chronic arthritis-retrospective study of 187 patients. Klinishe Pediatry. 198(5):401-7, 1986 Sap-Oct.
Prieur AM. Bremard-Oury C. Griscelli C. Mozziconacci P. Prognosis of the systemic forms of juvenile chronic arthritis. Apropos of 100 cases. Archives Francaises de Pédiatrie. 41(2):91-7. 1984 Feb.
Svantesson H, Akeson A, Eberhardt K, Elbourgh R. Prognosis in juvenile rheumatoid arthritis with systemic onset: a follow-up study. Scand J Rheumatology 12:139-44, 1983
Talesnik E. Leissner M. Jacobelli S. Juvenile systemic rheumatoid arthritis and adult-onset Still's disease: comparison of clinical course. Revista Modica de Chile. 120(6):638-43, 1992 Jun.
Tanaka S. Matsumoto Y. Ohnishi H. Moods M. Nishioka K. Kashiwazaki S. Watanabe N. Comparison of clinical features of childhood and adult onset Still’s disease. Ryumachi. 31(5):511-8. 1991 Oct.
History of AOSD
Ansell BM. Still's disease. Brit J Clin Pract 45:212-5, 1991
Bannatyne GA, Wohlman AS, Blaxall FR. Rheumatoid arthritis: its clinical history, etiology, and pathology. Lancet 1:1120-25, 1896
Baum J, Baum ER. George Frederic Still and his account of childhood arthritis - reappraisal. Am J Dis Child 132:192-4, 1978
Birch CA. Still's disease. George Frederick Still (1868-1941). Practitioner. 210(256):307-8, 1973 Feb.
Boldero MEA. A case of Still's disease. Mod Soc Trans London 16:55, 1933
Chauffard A, Ramond F. Rev de Med. 16:345, May 1896
Fanconi G. Uber einen fall von subsepsis allergica Wissler. HeIv Paediat 1: 532, 1946
MoItke 0. Still's disease in adults: a contribution to the symptomatology of subchronic polyarthritis. Acta Medica Scandinavia 80:427-53, 1933
Wissle H. Uber eine besondere form sepsisahnlicher Krankheiten (Subsepsis hyperallorgica). Mschr. Kinderheilk 94:51, 1943
Fever and AOSD
Aduan R. et al. Clinical Research 26:558A, 1978
Anonymous. Fever In Still's disease. Lancet. 1(500) 145-6. 1967
Cohen N. Weissgarten J. Golik A. Pik A. Yona E. Pallor S. Modai D. Adult Still's disease presenting as PUO; a not uncommon disorder. 5. British Journal of Rheumatology. 26(l):65-6, 1987 Feb.
Kazanjian PH. Fever of unknown origin: review of 86 patients treated in community hospitals. Clin Infect Dis 15:968-73, 1992
Kluger MJ. Fever: role of pyrogens and cryogens. Physiological Reviews. 710):93-127. 1991 Jan.
Knockaert DC, Vanneste U, Bobbaers HJ. Recurrent or episodic fever of unknown origin: review of 45 cases and survey of the literature. Medicine 72:184-96, 1993
Larson EB, Featherstone HJ, Petersdorf RG. Fever of undetermined origin: diagnosis and follow-up of 105 cases, 1970-80. Medicine 61:269-93, 1982
Mizock BA. Balamuniswarny G. Adult Stills disease presenting as favor of undetermined origin in a patient with leukemoid reaction. Western Journal of Medicine. 157(5):S73-5, 1992 Nov.
Smith JW. Favor of undetermined origin: not what it used to be. American Journal of the Medical Sciences. 292(1):56-64, 1986 Jul.
Still's (Rheumatoid) Rash
Bhanji AS. Bruckner FE. Adult onset Still's disease without a rash. Journal of the Royal Society of Medicine. 77(7):617-8, 1984 Jul.
Henry K. Adult Still's disease presenting with fever and a pruritic rash. Minnesota Medicine. 69(9):525-6, 1986 Sep.
Isdale IC, Bywaters EGL. The rash of juvenile rheumatoid arthritis. Quarterly J Medicine 23:377-387, 1956
Kaur S. Bambery P. Dhar S. Persistent dermal plaque lesions in adult onset Still's disease. Dermatology. 188(3):241-2. 1994.
Phillips WG. Weller R. Handfield-Jones SE. Kobza-Black A. Adult Still's disease. British Journal of Dermatology. 130(4):511-3, 1994 Apr.
Ridgway HA. Adult-onset Still's disease. Journal of the Royal Society of Medicine. 75(6):474-6. 1982 Jun.
Arthritis and Carpal Ankylosis
Cush JJ. Medsger TA Jr. Christy WC. Herbert DC. Cooperstain LA. Adult-onset Still's disease. Clinical course and outcome. Arthritis & Rheumatism. 30(2):186-94, 1987 Feb.
De Mulder PH. van de Putte BL. Adult-onset Still's disease: destructive distal interphalangeal arthritis associated with transient capsular calcification. Annals of the Rheumatic Diseases. 41(5):544-6, 1982 Oct.
Garcia-Morteo 0. Gusis SE. Somma LF. Maldonado-Cocco JA. Tarsal ankylosis in juvenile and adult onset . rheumatoid arthritis. Journal of Rheumatology. 15(2):298-300, 1988 Feb.
Medsger TA Jr. Christy WC. Carpal arthritis with ankylosis in late onset Still's disease. Arthritis & Rheumatism. 19(2):232-42, 1976 Mar-Apr.
Mozziconaci P. Prieur AM. Hayem F. Oury C. Joint prognosis in systemic chronic juvenile arthritis (100 cases). Semaine des Hopitaux. 59(48):3357-60, 1983 Dec 22.
Wouters JM. Froeling PG. van de Putte LS. Adult-onset Still’s disease complicated by hypercalcaemia: possible relationship with rapidly destructive polyarthritis. Annals of the Rheumatic Diseases. 44(5):345-8. 1985 May.
Zajaczek-Grabowska A. Maldykowa H. Chwalinska-Sadowska H. Ankylosis of carpal joints as a characteristic .radiological finding in adult cases of Still's disease. Reumatologia. 16(4):539-45, 1978.
Myopathy and Myocarditis in AOSD
Bank I. Marboe CC. Redborg RF. Jacobs J. Myocarditis in adult Still's disease. Arthritis & Rheumatism. 28(4):452-4, 1985 Apr.
Barbadillo, C. Cuende E. Mazzucchelli R. Mulero J. Andreu-Sanchez JL. Still's disease in adults and polymyositis. An infrequent association. Medicina Clinica. 99(10)382-3, 1992 Oct 3.
Hosaka S. Takashina N. Ishikawa A. Kondo H. Kashiwazaki S. Adult Still's disease with rnyocarditis and peritonitis. Internal Medicine. 31(6):812-5, 1992 Jun.
Moreno-Alvarez MJ. Citera G. Maldonado-Cocco JA. Taratuto AL. Adult Still's disease and inflammatory myositis. Clinical & Experimental Rheumatology. 11 (6):659-61, 1993 Nov-Dec.
Sachs RN. Talvard 0. Lanfranchi J. Myocarditis in adult Still's disease. International Journal of Cardiology. 27(3):377-80, 1990 Jun.
Samuels AJ. Barney SN. Tourtellotte CD. Artymyshyn R. Coexistence of adult onset Still's disease and polymyositis with rhabdomyolysis successfully treated with methotroxate and corticosteroids. Journal of Rheumatology. 16(5):685-7, 1989 May.
Schwarzberg C. Le Goff P. Le Menn G. Muscular involvement in adult Still's disease. Apropos of 5 now cases. Semaine des Hopitaux. 58(41):2408-12, 1982 Nov 11.
Ward SC, Wiselka MJ, Nicholson KG. Still's disease and myocarditis associated with recent mumps infection. Postgraduate Medical Journal 64:693-95, 1988
Lymphadenopethy, Lymph Node Biopsy, Lymphoma and Hemphagocytic Syndrome
Anonymous. Lymphodenopathy in Still's disease of the adult. Medicine Clinica. 82(11):515-6, 1984 Mar 24.
Coffernils M. Soupart A. Pradier 0. Feremans W. Neve P. Decaux G. Hyperferritinemia in adult onset Still's disease and the hemophagocytic syndrome. Journal of Rheumatology. 19(9):1425-7, 1992 Sep.
Lyberatos C. Two more cases of Still's disease and Kikuchi's. J Rheumatology 17:568-9, 1990
Kawasaki T, Hirohata S, Hashimoto T, Miyashita H, Tanaka F. T cell lymphoma in adult Still’s disease. Clin Exp Rheumatol (in press), 1995
McKenna RW, Risdall RJ, Brunning RD. Virus associated hemophagocytic syndrome. Human Pathology 12:395-98, 1981
McPeake JR, Hirst WJR, Brind AM, Williams R. Hepatitis A causing a second episode of virus-associated haemophagocytic lymphohistiocytosis in a patient with Still's disease. Journal Medical Virology 39:173-75, 1993
Morris JA, Adamson AR, Holt PJL, Davson J. Still's disease and the virus-associated haemophagocytic syndrome
Ohta A, Matsumoto Y, Ohta T, Kaneoka H, Yamaguchi M. Still's disease associated with necrotizing lymphadenitis (Kikuchi's disease): Report of 3 cases. J Rheumatology 15:981-83, 1991
Quaini F. Manganelli P. Pileri S. Magnani G. Ferrari C. Delsignore R. Sabattini E. Olivetti G. Immunohistological characterization of lymph nodes in two cases of adult onset Still's disease. Journal of Rheumatology. 18(9):1418-23, 1991 Sep.
Reichert U. Keuning JJ. van Seek M. van Rijthoven AW. Lymph node histology simulating T-cell lymphoma in adult-onset Still’s disease. Annals of Hematology. 650):53-4, 1992 Jul.
Trotta F. Dovigo, L. Scapoli G. Cavazzini L. Castoldi G. Immunoblastic malignant lymphoma in adult onset Still's disease. Journal of Rheumatology. 20(10)788-92, 1993 Oct.
Valente RM. Banks PM. Conn DL. Characterization of lymph node histology in adult onset Still's disease. Joumal of Rheumatology. 16(3):349-54, 1989 Mar.
Pregnancy and AOSD
De Miguel E. Cuesta M. Martin-Mola E. Gijon-Banos J. Adult Still’s disease and pregnancy. Journal of Rheumatology. 19(3):498, 1992 Mar.
Green J. Kanter Y. Barzilai D. Adult Still's disease associated with pregnancy. Israel Journal of Medical Sciences. 18(10):1037-9, 1982 Oct.
Katz WE. Starz TW. Winkelstein A. Recurrence of adult Still's disease after pregnancy. Journal of Rheumatology. 17(3):373-4. 1990 Mar.
Le Loet X. Daragon A. Duval C. Thomine E. Lauret P. Humbert G. Adult onset Still's disease and pregnancy. Journal of Rheumatology. 20(7):1158-61, 1993
Stain GH. Cantor B. Panush RS. Adult. Still's disease associated with pregnancy. Arthritis & Rheumatism. 23(2):248-50, 1980 Feb.
Yebra Bango M. Garcia Paez JM. Solovera JJ. Merino MF. Giron Gonzalez JA. Adult-onset Still's disease: a case with onset during pregnancy. Arthritis & Rheumatism. 28(8):957, 1985 Aug.
Uncommon Associations and Complications with AOSD
Aellen P. Raccaud 0. Waldburger M. Chamot AM. Gerster JC. Still's disease in adults with disseminated intravascular coagulation. Schweizerische Rundschau fur Medizin Praxis. - 80(15):376-8, 1991 Apr 9.
Alukal MK. Costello PB. Green FA. Cardiac tamponade in systemic juvenile rheumatoid arthritis requiring emergency pericardiectomy. Journal of Rheumatology. 11 (2):222-5, 1984 Apr.
Bosch X. Gonzalez-Clemente JM. Cervera R. Grau-Junyent JM. Sjogren's disease with adult onset Still's disease. Journal of Rheumatology. 16(6):847-8, 1989 Jun.
Braidy JF. Adult onset Still's disease. Chest. 940):221-2, 1988 Jul.
Braidy JF. Poulson JM. Diaphragmatic weakness and myositis associated with systemic juvenile rheumatoid arthritis. Canadian Medical Association Journal. 1300):47-9, 1984 Jan 1.
Brandwein SR. Salusinsky-Sternbach M. Adult Still’s disease in only one of identical twins. Journal of Rheumatology. 16(12):1599-601, 1989 Dec.
Bruckle W. Eisenhut C. Goebel FD. Cerebral involvement in adult onset Still's disease. Clinical Rheumatology. 11 (2):276-9, 1992 Jun.
Cantor JP. Pitcher WO. Hurd E. Severe restrictive pulmonary defect in a patient with adult-onset Still's, disease. Chest. 92(5):939-40, 1987 Nov.
Cook PJ. Adult Still's disease with late relapse. Proceedings of the Royal Society of Medicine. 890):60, 1976 Jan.
Corbett AJ. Zizic, TM. Stevens MEL Adult-onset Still's disease with an associated severe restrictive pulmonary defect: a case report. Annals of the Rheumatic Diseases. 42(4):452-54, 1983 Aug.
Cush JJ. Leibowitz lH. Friedman SA. Adult-onset Still's disease and inflammatory orbital pseudotumor. Now York State Journal of Medicine. 85(3):110-1. 1985 Mar.
De Bandt M. Kahn MF. Takayasu’s arteritis associated with Still's disease in an adult.. Clinical & Experimental Rheumatology. 9(6):639-40, 1991 Nov-Dec.
Donault A. Dimopoulos MA. Fitzcharies MA. Meningoencephalitis and peripheral neuropathy complicating adult Still's disease. Journal of Rheumatology. 17(5):698-700, 1990 May.
Drouot MH. Hachulla E. Flipo RM. Hatron PY. Houvenagel E. Gosset D. Ducloux G. Devulder S. Cardiac complication of adult-onset Still's disease: from pericarditis to tamponade, sometimes a manifestation of the disease. Revue de Medecine Interne. 14(10):1017, 1993.
Garrote FJ. Marco J. Obeso G. Rodriguez E. del Ser T. Aseptic meningitis and focal central nervous system involvement in a case of adult onset Still's disease. Journal of Rheumatology. 20(4):765-7, 1993 Apr.
Gibbs CJ. Foord C. Lee HA. Smith G. Disseminated intravascular coagulation in adult-onset Still's disease with neurological, respiratory and hepatic sequelae. British Journal of Hospital Medicine. 50(5):278-9, 1993 Sep 1-14.
Goldenberg J. Pessoa AP. Roizenblatt S. Pavoa RM. Hilario MO. Atra E. Ferraz MB. Cardiac tamponade in juvenile chronic arthritis: report of two cases and review of publications. Annals of the Rheumatic Diseases. 49(7):549-53, 1990 Jul.
Harrington TM. Moran JJ. Davis DE. Amyloid9sis in adult onset Still's disease. Journal of Rheumatology. 8(5):833-6, 1981 Sep-Oct.
Hirohata S. Kamoshita H. Taketani T. Maeda S. Adult Still’s disease complicated with adult respiratory distress. Archives of Internal Medicine. 146(12):2409-10, 1986 Dec.
Jamieson TW. Adult Still's disease complicated by cardiac tamponade. JAMA. 249(15):2065-8, 1983 Apr 15.
Kahl LE. Aplastic anemia in adult onset Still’s disease during gold therapy. Journal of Rheumatology. 15(12):1877-8, 1988 Dec.
Kaplinsky N. Pras M. Franki 0. An adult form of juvenile rheumatoid arthritis. Archives of Internal Medicine. 140(8):1073-4, 1980 Aug.
Kaufman LD. Sibony PA. Anand AK. Gruber BL. Superior oblique tenosynovitis (Brown's syndrome) as a manifestation of adult Still’s disease. Journal of Rheumatology. 14(3):625-7, 1987 Jun.
Koga T. Tokunaga N. Ichikawa Y. Oizumi K. A 72-year-old female with adult Still's disease. Internal Medicine. 31(12):1356-8, 1992 Dec.
Liote H. Liote F. Lonique F. Milleron BJ. Kuntz D. Akoun GM. Adult-onset Still's disease revealed by a pleuropericarditis Apr;4(4):513. European Respiratory Journal. 3(9):1064-6, 1990 Oct.
Markusse HM. Stolk B. van der Mey AG. do Jonge-Bok JM. Heering KJ. Sensorineural hearing loss in adult onset Still's disease. Annals of the Rheumatic Diseases. 47(7):600-2. 1988 Jul.
Pedersen JE. ARDS-associated with adult Still's disease. Intensive Care Medicine. 17(6):372, 1991.
Pollet SM, Vogt PJ, Leek JC. Serous peritonitis in adult Still's syndrome. Journal of Rheumatology 17:98-101, 1990
Rossor E. Takayasu's arteritis as a differential diagnosis of systemic juvenile chronic arthritis. Archives of Disease in Childhood. 54(10):798-800, 1979 Oct.
Schwartz D. Averbuch M. Pines A. Kornovsky R. Levo Y. Disseminated intravascular coagulation with renal and liver damage as the predominant manifestations of recurrent relapses in systemic juvenile rheumatoid arthritis. Annals of the Rheumatic Diseases. 51(3):347-9, 1992 Mar.
Sherry DD. Kredich DW. Transient thrombocytopenia in systemic onset juvenile rheumatoid arthritis. Pediatrics. 76(4):600-3, 1985 Oct.
Sibley JT. Concurrent onset of adult onset Still's disease and insulin dependent diabetes mellitus. Annals of the Rheumatic Diseases. 49(7):547-8, 1990 Jul.
Silverman ED. Miller JJ 3d. Bernstein S. Shafai T. Consumption coagulopathy associated with systemic juvenile rheumatoid arthritis. Journal of Pediatrics. 103(6):872-6, 1983 Dec.
Sugawara T. Tsukada T. Wakita Y. Wake Y. Kouyama K. Tamaki S. Tanigawa M. Iwasaki E. Ohta C. Kageyana S. et al. A case of myelodysplastic syndrome progressing to acute myelocytic leukemia in which adult-onset Still's disease had occurred 6 years before. International Journal of Hematology. 59(l):53-7, 1993 Dec.
Uson J. Pena JM. del Arco A. Barbado FJ. Vazquez JJ. Still's disease in a 72-year-old man. Journal of Rheumatology. 20(9):1608-9, 1993 Sep.
Van Hoeyweghen RJ. De Clerck LS. Van Offel JF. Stevens WJ. Interstitial lung disease and adult-onset Still's disease. Clinical Rheumatology. 12(3):418-21, 1993 Sep.
Wendling D. Humbert PG. Billerey C. Fast T. Dupond JL. Adult onset Still's disease and related renal amyloidisis. Annals of the Rheumatic Diseases. 50(4):257-9, 1991 Apr.
Wilson WA. Morgan OS. Bain B. Taylor JE. Takayasu's arteritis: association with Still's disease in an adult. Arthritis & Rheumatism. 22(6):684-8, 1979 Jun.
Wouters JM. van Rijswijk MH. van de Putte LB. Adult onset Still’s disease in the elderly: a report of two cases. Journal of Rheumatology. 12(4):791-3, 1985 Aug.
Hyperferritinemia in AOSD
Coffernils M. Soupart A. Pradier 0. Feremans W. Neve P. Decaux G. Hyperferritinernia in adult onset Still's disease and the hemophagocytic syndrome. Journal of Rheumatology. 19(9):1425-7, 1992 Sep.
Gonzalez-Hernandez T. Martin-Mola E. Fernandez-Zamorano A. Balsa-Criado A. de Miguel-Mandieta E. Serum ferritin can be useful for diagnosis in adult onset Still's disease. Journal of Rheumatology. 16(3):412-3. 1989 Mar.
Olive A. Junca J. Tena X. Ferritin and adult Still's disease. British Journal of Rheumatology. 30(2):158, 1991 Apr.
Ota T. Higashi S. Suzuki H. Eto S. Increased serum ferritin levels in adult Still's disease. Lancet. (8532):562-3, 1987 Mar 7.
Pelkonen P. Swanljung K. Siimes MA. Ferritinemia as an indicator of systemic disease activity in children with systemic juvenile rheumatoid arthritis. Acta Paediatrica Scandinavica. 75(1):64-8, 1988 Jan.
Rogers JT, Bridges KR, Durmowicz GP, Glass J, Auron PE, Munro HN. Translational control during the acute phase response. J Biol Chem 265:14572-78, 1990
Sanmarti R. Toribio R. Panella D. Lopez de Aguileta A. Serum ferritin in adult Still’s disease. Medicine Clinics. 94(9):357-8. 1990 Mar 10.
Schwarz-Eywill M. Heilig B. Bauer H. Breitbart A. Pezzutto A. Evaluation of serum ferritin as a marker for adult Still's disease activity. Annals of the Rheumatic Diseases. 51 (5):683-5, 1992 May.
Tsuji Y, Miller LL, Miller SC, Torti SV, Torti FM. Tumor necrosis factor-a and interleukin 1-a regulate transferrin receptor in human diploid fibroblasts. J Biol Chem 266:7257, 1991
Van Reeth C. Le Moel G. Lasne Y. Revenant MC. Agneray J. Kahn MF. Bourgeois P. Serum ferritin and isoferritins are tools for diagnosis of active adult Still’s disease. Journal of Rheumatology. 21(5):890-5. 1994 May.
Infectious Associations with AOSD
Balleari E. Cutolo M. Accardo S. Adult-onset Still's disease associated to toxoplasma gondii infection. Clinical Rheumatology. 10(3):326-7, 1991 Sep.
Chantler JK, Tingle AJ, Petty RE. Persistent rubella virus infection associated with chronic arthritis in children. New England J Medicine 313:1117-23, 1985
Cimmino MA. Trevisan G. Lyme arthritis presenting as adult onset Still's disease. Clinical & Experimental Rheumatology. 7(3):305-8, 1989 May-Jun.
Colebunders R. Stevens WJ. Vanagt E. Snoeck J. Adult Still's disease caused by Yersiria enterocolitica infection. Archives of Internal Medicine. 144(9):1880-2, 1984 Sep.
De Vere-Tyndall A. Bacon T. Parry R. Tyrrell DA. Denman AM.Ansell BM. Infection and interferon production in systemic juvenile chronic arthritis: a prospective study. Annals of the Rheumatic Diseases. 43(l):1-7, 1984 Feb.
Gordon SC. Lauter CS. Mumps arthritis: unusual presentation as adult Still's disease. Annals of Internal Medicine. 97(1):45-7. 1982 Jul.
Grahame R. Armstrong R. Simmons N.. Wilton JM. Dyson M. Laurent R. Millis R. Mims CA. Chronic arthritis associated with the presence of intrasynovial rubella virus. Annals of the Rheumatic Diseases. 42(1):2-13, 1983 Feb.
Guzzini F. Banfi L. Camerone, G. Florio G. Garelli S. Cold agglutinin hemolytic anemia in a patient with adult-onset Still's disease. Haematologica. 75(5):467-9, 1990 Sep-Oct.
Howard A. Ansell SM. Powell R. Proceedings: Immunoglobulin levels during rubella in children suffering from Still's disease. Annals of the Rheumatic Diseases. 34(2):204, 1975 Apr.
Huang SH. DeCoteau WE. Adult-onset Still's disease: an unusual presentation of rubella infection. Canadian Medical Association Journal. 122(11):1275-6, 1980 Jun 7
Jones JG. Adult onset Still's disease or coxsackie polyarthritis? Australian & Now Zealand Journal of Medicine. 17(2):259, 1987 Apr.
Karjalainen J. Heikkila J. Nieminen MS. Jalanko H. Kleemola M. Lapinleimu K. Sahi T. Etiology of mild acute infectious myocarditis. Relation to clinical features. Acta Medica Scandinavica. 213(1):65-73, 1983.
Lang DM. Petrozzi C. Immune complex disease--adult Still's disease? Hepatitis B?. Western Journal of Medicine. 143(5):680-1, 1985 Nov.
Newkirk MM. Lemmo A. Commerford K. Esdaile JM. Brandwein S. Aberrant cellular localization of rubella viral genome in patients with adult Still's disease--a pilot study. Autoimmunity. .16(1)39-43, 1993.
Newport M. Conway SP. Huston G. A case of cytomegalovirus demonstrating the need for a protocol of infection screening when classifying adult Still's disease. Journal of Rheumatology. 20(4):757-8,, 1993 Apr.
Ogra PL, Ogra SS, Chiba Y, Dzierba JL. Rubella-virus infection in juvenile rheumatoid arthritis. Lancet i:1157-61, 1975
Pouchot J. Ouakil H. Debin ML. Vinceneux P. Adult Still's disease associated with acute human parvovirus B19 infection. Lancet. 341(8855):1280-1, 1993 May 15.
Rahal JJ, Milian SJ, Noriega ER. Coxackie and adenovirus infection: association with acute febrile and juvenile arthritis. JAMA 235:2496-2501, 1976
Roberts-Thomson PJ. Southwood TR. Moore BW. Smith MO. Ahern MJ. Geddes RA. Hill WR. Adult onset Still's disease or coxsackie polyarthritis?. Australian & New Zealand Journal of Medicine. 16(4):509-11, 1986 Aug.
Ward SC, Wiselka IAJ, Nicholson KG. Still's disease and myocarditis associated with recent mumps infection. Postgraduate Medical Journal 64:693-95, 1988
Wouters, JM. van der Veen J. van de Putte LB. de Rooij DJ. Adult onset Still’s disease and viral infections. Ann Rheum Dis 47:764-67, 1988
Immunologic Abnormalities in Still's Disease
Bacon TH. de Vere-Tyndall A. Tyrrell DA. Denman AM. Ansell BM. Interferon system in patients with systemic juvenile chronic arthritis: in vivo and in vitro studies. Clinical & Experimental Immunology. 540):23-30, 1983 Oct.
Bedford PA, Ansell SM, Hall PJ, Woo P. Increased frequency of DR4 in systemic onset juvenile chronic arthritis. Clinical and Experimental Rheumatology 10:189-93. 1992
Miller ML. Aaron S. Jackson J. Fraser P. Cairns L. Hoch S. Sorel Y. Larson M. Glass DN. HLA gene frequencies in children and adults with systemic onset juvenile rheumatoid arthritis. Arthritis & Rheumatism. 28(2):146-50, 1985 Feb.
Prieur AM. Kaufmann MT. Griscelli C. Dayer JM. Specific interleukin-1 inhibitor in serum and urine of children with systemic juvenile chronic arthritis. Lancet. 2(8570):1240-2, 1987 Nov 28.
Reidbord HE. Osial TA Jr. Synovial lymphocyte subsets in rheumatoid arthritis and degenerative joint disease. Journal of Rheumatology. 14(6):1089-94, 1987 Dec.
Terkeltaub R. Esdaile JM. Decary F. Harth M. Lister J. Lapointe N. HLA-Bw35 and prognosis in adult Still’s disease. Arthritis & Rheumatism. 24(12)1469-72, 1981 Dec.
Tsokos GC. Mavridis A. Inghirami G. Pillemer SR. Emery HM. Magilavy DB. Cellular immunity in patients with systemic juvenile rheumatoid arthritis. Clinical Immunology & Immunopathology. 420):86-92, 1987 Jan.
Wouters JM. Reekers P. van de Putte LB. Adult-onset Still’s disease. Disease course and HLA associations. Arthritis & Rheumatism. 29(3):415-8, 1986 Mar.
Diagnosis of AOSD
Calabro JJ. Londino AV Jr. Adult onset Still's disease. Journal of Rheumatology. 13(4):827-8, 1986 Aug.
Cush JJ. Adult-onset Still's disease. Rheumatology Grand Rounds, University of Pittsburgh Medical Center; Jan.30, 1984
Murden RA. Adult-onset Still's disease with a significant rheumatoid factor: examining proper use of diagnostic and classification criteria. Journal of General Internal Medicine. 7(3):366-8, 1992 May-Jun.
Ostrov BE. Goldsmith DP. Athreya SH. Differentiation of systemic juvenile rheumatoid arthritis from acute leukemia near the onset of disease. Journal of Pediatrics. 122(4):595-8, 1993 Apr.
Reginato AJ. Schumacher HR Jr. Baker DG. O'Connor CR. Ferreiros J. Adult onset Still's disease: experience in 23 patients and literature review with emphasis on organ failure. Seminars in Arthritis & Rheumatism. 17(1)39-57, 1987 Aug.
Yamaguchi M. Ohts- A. Tsunematsu T. Kasukawa R. Mizushima Y. Kashiwagi H. Kashiwazaki S. Tanimoto K. Matsumoto Y. Ota T. et al. Preliminary criteria for classification of adult Stilt's disease. Journal of Rheumatology. 19(3):424-30, 1992 Mar.
Therapy In AOSD
Aydintug AO. D’Cruz D. Cervera R. Khamashta MA. Hughes G.R. Low dose methotrexate treatment in adult Still's disease. Journal of Rheumatology. 19(3):431-5, 1992 Mar.
Bisagni-Faure A. Job-Deslandre C. Menkes CJ. Treatment of Still's disease with bolus methylprednisolone. Retrospective study of 7 patients. Revue du Rhumatisme et des Maladies Osteo-Articulaires. 59(3):228-32, 1992 Mar.
Bjerkhoel F. Forre 0. Cyclosporin treatment of a patient with severe systemic juvenile rheumatoid arthritis. Scandinavian Journal of Rheumatology. 17(6):483-6, 1988.
Bliddal H. Helin P. Leucopenia in adult Still's disease during treatment with azathioprine and sulphasalazine. Clinical Rheumatology. 6(2):244-50, 1987 Jun.
Crowley J. Situnayake RD. Sulphasalazine induced hepatitis in adult Still's disease. Annals of the Rheumatic Diseases. 51(11):1264-5, 1992 Nov.
Groothoff JW. van Leeuwen EF. High dose intravenous gammaglobulin, in chronic systemic juvenile arthritis. British Medical Journal - Clinical Research. 296(6633):1362-3, 1988 May 14.
Kenouch S. Fessi H. Mery JP. Belmatoug N. Kahn MF. Oral low-dose methotrexate (MTX) to be efficient in refractory rheumatoid arthritis and adult-onset Still's disease. American Journal of Kidney Diseases. 14(1):76-7, 1989 Jul.
Khraishi M. Fam AG. Treatment of fulminant adult Still's disease with intravenous pulse methylprednisolone therapy. Journal of Rheumatology. 18(7):1088-90, 1991 Jul.
Kraus A. Alarcon-Segovia D. Fever in adult onset Still's disease. Response to methotrexate. Journal of Rheumatology. 18(6):918-20, 1991 Jun.
Perrice W. Schuchmann L. Dippell J. Suschke J. Vogel P. Truckenbrodt H. Schindera F. Humburg C. Brzoska J. Therapy for systemic juvenile rheumatoid arthritis with gamma- interferon: a pilot study of nine patients. Arthritis & Rheumatism. 32(5):643-6, 1989 May.
Sato M. Takeda A. Honzu H. Saku N. Minato N. Kano S. Adult Still's disease with Sjogren's syndrome successfully treated with intravenous pulse methylprednisolone and oral cyclophosphamide. Internal Medicine. 32(9):730-2, 1993 Sep.
Shaikov AV. Maximov AA. Speransky Al. Lovell DJ. Giannini EH.Solovyev SK. Repetitive use of pulse therapy with methylprednisolone and cyclophosphamide in addition to oral methotroxate in children with systemic juvenile rheumatoid arthritis--preliminary results of a longterm study. Journal of Rheumatology. 19(4):612-6, 1992 Apr.
Silverman ED. Laxer RM. Greenwald M. Gelfand E. Shore A. Stein LD. Roifman CM. Intravenous gamma globulin therapy in systemic juvenile rheumatoid arthritis. Arthritis & Rheumatism. 33(7):1015-22, 1990 Jul.
Hollingworth P. Craft A. Ansell BM. Low-dose methotroxate in systernic onset juvenile chronic arthritis. Clinical & Experimental Rheumatology. 7(6):647-50, 1989 Nov-Dec.
Circadian, Cytokine Hypothesis
Arvidson NG, Gudbiornsson B, Elfman L, Ryden AC, Totterman, TH, Hallgren R. Circadian rhythmn of serum interleukin-6 in rheumatoid arthritis. Ann Rheum Dis 53:521-24, 1994
Covelli V. Massari F. Fallacara C. Munno I. Jirillo E. Savastano S. Tommasalli AP. Lombardi G. Interleukin-1 beta and beta-endorphin circadian rhythms are inversely related in normal and stress-altered sleep. International Journal of Neuroscience. 63(3-4):299-305, 1992 Apr.
David J, Davies U, Rooney M. Cytokine measurement in juvenile chronic arthritis (JCA). Arthritis Rheum 33[suppl]:S 118, 1990
De Benedtti F. Massa M. Pignatti P. Albani S. Novick D. Martini A. Serum soluble interleukin 8 (IL-8) receptor and IL-6/soluble IL-6 receptor complex in systemic juvenile rheumatoid arthritis. Journal of Clinical Investigation. 93(5):2114-9, 1994 May.
De Benedetti F. Massa M. Robbioni P. Ravelli A. Burgio GR. Martini A. Correlation of serum interleukin-6 levels with joint involvment and thrombocytosis in systemic juvenile rheumatoid arthritis. Arthritis & Rheumatism. 34(9):1158-63, 1991 Sep.
Gudewill S. Poilmacher T. Vedder H. Schreiber W. Fassbender K. Holsboer F. Nocturnal plasma levels of cytokines in healthy men. European Archives of Psychiatry & Clinical Neuroscience. 242(l):53-6, 1992.
Hrushesky WJ. Langevin T. Kim YJ. Wood PA. Circadian dynamics of tumor necrosis factor alpha (cachectin) lethality. Journal of Experimental Medicine. 180(3):1059-65, 1994 Sep 1.
Lernmer B. Schwulera U. Thrun A. Lissner R. Circadian rhythm of soluble interleukin-2 receptor in healthy individuals. European Cytokine Network. 3(3):335-6, 1992 May-Jun.
Neeck G. Federlin K. Graef V. Rusch D. Schmidt KL. Adrenal secretion of cortisol in patients with rheumatoid arthritis. Journal of Rheumatology. 170):24-9, 1990 Jan.
Zabel P. Horst HJ Kreiker C. Schlaak M. Circadian rhythm of interleukin-1 production of monocytes and the influence of endogenous and exogenous, glucocorticoids in man. Klinische Wochenschrift. 68(24):1217-21, 1990 Dec 17.